大黃素通過減少DNMTs的表達(dá)對胰腺癌細(xì)胞抑癌基因ppENK發(fā)揮去甲基化作用研究
2023-11-13 05:45:50陳亮唐堅褚永權(quán)葉劍宏沈超錢曉宇
中國現(xiàn)代醫(yī)生 2023年30期
陳亮,唐堅,褚永權(quán),葉劍宏,沈超,錢曉宇
大黃素通過減少DNMTs的表達(dá)對胰腺癌細(xì)胞抑癌基因ppENK發(fā)揮去甲基化作用研究
陳亮,唐堅,褚永權(quán),葉劍宏,沈超,錢曉宇
嘉興市第一醫(yī)院頭頸外科,浙江嘉興 314000
研究大黃素對胰腺癌細(xì)胞Panc1 DNA甲基轉(zhuǎn)移酶(DNA methyltransferases,DNMTs)的影響,及其對抑癌基因前腦啡肽原(pre-proenkephalin,ppENK)啟動子區(qū)CpG島的去甲基化作用,探討大黃素是否可通過降低DNMTs的表達(dá)逆轉(zhuǎn)ppENK的甲基化狀態(tài)。CCK-8檢測不同濃度大黃素對Panc1細(xì)胞生長的影響,探索最佳用藥濃度,重亞硫酸鹽測序PCR(bisulfite genomic sequencing PCR,BSP)檢測不同濃度大黃素對ppENK基因甲基化狀態(tài)的影響,PCR和Western Blot檢測ppENK及甲基轉(zhuǎn)移酶DNMT1、DNMT3a的mRNA和蛋白表達(dá)情況。大黃素以時間梯度和濃度梯度依賴性抑制Panc1細(xì)胞生長,其半數(shù)最大抑制濃渡(half maximal inhibitory concentration,IC50)約為40μmol/L,大黃素可使ppENK甲基化狀態(tài)減弱,非甲基化狀態(tài)增強(qiáng);可減弱DNMTs的mRNA和蛋白表達(dá),增強(qiáng)ppENK mRNA和蛋白表達(dá)。大黃素可不同程度地使Panc1 ppENK抑癌基因去甲基化,使抑癌基因重新表達(dá)。大黃素抑制胰腺癌細(xì)胞生長和其去甲基化作用有關(guān),推測大黃素發(fā)揮去甲基化作用和其抑制甲基轉(zhuǎn)移酶表達(dá)相關(guān)。
大黃素;胰腺癌;去甲基化;DNA甲基轉(zhuǎn)移酶;重亞硫酸鹽測序PCR
胰腺癌是一種常見的惡性腫瘤,具有侵襲性強(qiáng)、早期癥狀不典型、術(shù)后易轉(zhuǎn)移復(fù)發(fā)等特點。據(jù)國際癌癥研究機(jī)構(gòu)統(tǒng)計,胰腺癌的死亡率居第7位,發(fā)病率居36種常見惡性腫瘤的第14位[1]。另一項統(tǒng)計數(shù)據(jù)表明,美國2021年新增胰腺癌確診病例60 430例,死亡病例48 220例,到2030年胰腺癌或?qū)⒊蔀槊绹┌Y相關(guān)死亡的第二大原因[2-3]。
腫瘤發(fā)生、發(fā)展過程中的表觀遺傳修飾變化備受關(guān)注,表觀遺傳修飾主要包括:DNA甲基化和羥甲基化、染色質(zhì)重塑、組蛋白甲基化和乙酰化、RNA干擾、基因組染色等。催化DNA甲基轉(zhuǎn)移酶(DNA methyltransferases,DNMTs)有DNMT1、DNMT3a和DNMT3b,進(jìn)而形成5mC[4]。DNA甲基化修飾與轉(zhuǎn)錄失活、基因印記、X染色體失活密切相關(guān),大量研究證據(jù)表明,DNA多重甲基化在多種惡性腫瘤的發(fā)生、發(fā)展中起重要作用[5]。
研究發(fā)現(xiàn)在胰腺癌中,許多抑癌基因存在甲基化異常,包括CDKN1C、SPARC、P16、RASSF1A和ppENK等,都伴隨著不同程度的甲基化,而其mRNA表現(xiàn)出不同程度的表達(dá)缺失[6-10]。這些抑癌基因中,以ppENK研究相對比較明確。Ueki等[10]和Fukushima等[11]報道超過90%的胰腺癌ppENK基因甲基化水平上升。
既往研究發(fā)現(xiàn)大黃素可抑制胰腺癌細(xì)胞生長,致基因組5mC水平下降,使抑癌基因ppENK的甲基化水平下降,非甲基化水平上升,本文是后續(xù)研究,探討大黃素對Panc1 DNMTs的影響,對ppENK的去甲基化作用,探討大黃素是否可通過降低DNMTs的表達(dá)逆轉(zhuǎn)ppENK的甲基化狀態(tài),從而使其重新表達(dá)生物活性(mRNA和蛋白水平),這一發(fā)現(xiàn)將為大黃素的臨床抗腫瘤治療奠定理論基礎(chǔ)。
1 材料與方法
1.1 材料
大黃素、5AzA-cdR購自Sigma公司,CCK-8試劑盒購自Gibco公司。細(xì)胞/組織基因組DNA提取試劑盒(離心菌株型)購自上海杰瑞生物工程有限公司,EpiTect?bisulfite kit改良試劑盒和ppENK抗體購自Abcam公司。
1.2 方法
1.2.1 細(xì)胞系和培養(yǎng) 本研究引進(jìn)并保藏人胰腺癌細(xì)胞系Panc1細(xì)胞株,培養(yǎng)于含胎牛血清、鏈霉素和青霉素的DMEM培養(yǎng)基中,每2~4d更換一次培養(yǎng)基,當(dāng)生長到70%~80%時傳代進(jìn)行實驗。
1.2.2 細(xì)胞增殖實驗(CCK-8) 取對數(shù)生長期的Panc1細(xì)胞,分別使用濃度為0μmol/L、10μmol/L、20μmol/L、40μmol/L、80μmol/L的大黃素處理細(xì)胞24h、48h和72h。用酶標(biāo)儀測定各孔在450nm波長下的吸光度(optical density,OD)值,實驗重復(fù)3次。計算細(xì)胞抑制率:細(xì)胞抑制率(%)=(1–實驗組OD值/對照組OD值)×100%。實驗組為含有細(xì)胞的培養(yǎng)基、CCK-8及濃度為10μmol/L、20μmol/L、40μmol/L、80μmol/L的大黃素處理組;對照組為含有細(xì)胞的培養(yǎng)基、CCK-8組,每組3個副孔。
1.2.3 DNA的提取與亞硫酸鹽修飾 使用細(xì)胞/組織基因組DNA提取試劑盒按說明書分別提取DNA,檢查DNA的純度和濃度后,取1μg DNA進(jìn)行亞硫酸鹽修飾,修飾過程按亞硫酸鹽修飾試劑盒說明書進(jìn)行。
1.2.4 焦磷酸鹽測序PCR(bisulfite genomic sequencing PCR,BSP) 提取各組DNA,取1μg DNA進(jìn)行亞硫酸鹽修飾,修飾過程參照EpiTect?bisulfite kit modification kit說明書進(jìn)行。取10μl PCR產(chǎn)物進(jìn)行凝膠電泳,結(jié)束后檢查BSP反應(yīng)產(chǎn)物,回收后送上海麥普生物技術(shù)有限公司進(jìn)行克隆,使用BiQ Analyzer軟件分析樣本的甲基化狀態(tài)。
1.2.5 聚合酶鏈反應(yīng)(polymerase chain reaction,PCR) 提取細(xì)胞總RNA,按RNA逆轉(zhuǎn)錄試劑盒說明書逆轉(zhuǎn)錄合成cDNA,制備10μl反應(yīng)體系,加一個標(biāo)準(zhǔn)孔和兩個副孔,上機(jī)(羅氏公司)擴(kuò)增45個循環(huán),結(jié)果用LightCycler480軟件分析,獨立重復(fù)實驗3次。
1.2.6 Western Blot 用RIPA提取各組蛋白,按BCA試劑盒說明書測定蛋白濃度,配制50μg體系,經(jīng)電泳、轉(zhuǎn)膜、封閉,4℃孵育各種濃度、種類的一抗,GAPDH作為內(nèi)參,以目的蛋白灰度值與GAPDH灰度值的比值作為衡量蛋白表達(dá)差異的參考,實驗重復(fù)3次。
1.3 統(tǒng)計學(xué)方法

2 結(jié)果
2.1 大黃素對胰腺癌細(xì)胞Panc1的生長抑制作用
本研究用濃度為0μmol/L、10μmol/L、20μmol/L、40μmol/L和80μmol/L的大黃素處理Panc1細(xì)胞24h、48h、72h(圖1),發(fā)現(xiàn)大黃素以時間和濃度梯度形式抑制Panc1細(xì)胞的生長,結(jié)果與Sheng等[12]研究類似。40μmol/L濃度處理Panc1細(xì)胞72h后,生長抑制率為49.4%;80μmol/L濃度時其抑制率達(dá)到64.4%。鑒于低濃度藥物不會對細(xì)胞產(chǎn)生毒副作用,故大黃素的實驗用藥濃度為0μmol/L、10μmol/L、20μmol/L和40μmol/L,用于后續(xù)研究。
2.2 BSP
為驗證上述結(jié)果的可靠性,筆者進(jìn)一步用BSP方法檢測ppENK的甲基化水平,BSP測序結(jié)果,見圖2。0μmol/L、10μmol/L、20μmol/L和40μmol/L的大黃素均可致ppENK不同程度去甲基化。ppENK基因測序范圍包括11個CpG島,隨機(jī)選取10個用于克隆,各組的大黃素分別作用于Panc1細(xì)胞72h后,ppENK基因甲基化率分別為86.4%、82.7%、67.3%、52.7%,5Aza-cdR組ppENK的甲基化率達(dá)39.1%。該結(jié)果進(jìn)一步證明大黃素可使ppENK的甲基化水平下降。

圖1 CCK-8實驗檢測結(jié)果

圖2 BSP實驗結(jié)果
注:黑色圓圈代表甲基化,空白圓圈代表非甲基化;*<0.05,#<0.01
2.3 大黃素對ppENK和DNMTs轉(zhuǎn)錄水平的影響
根據(jù)上述結(jié)果,大黃素、5AzA-cdR使ppENK的甲基化水平不同程度的降低,進(jìn)一步檢測了其對ppENK和DNMTs轉(zhuǎn)錄水平的影響。大黃素和1μmol/L 5AzA-cdR均能不同程度地增加ppENK的表達(dá),且5AzA-cdR的作用強(qiáng)于大黃素,同時大黃素、5AzA-cdR可不同程度地降低DNMT1和DNMT3a的表達(dá),見圖3。
2.4 大黃素對ppENK和DNMTs的蛋白水平的影響
根據(jù)上述結(jié)果,大黃素可不同程度地增強(qiáng)ppENK和DNMTs的mRNA表達(dá),本研究用Western Blot檢測是否有相應(yīng)的蛋白重新表達(dá),見圖4。結(jié)果表明大黃素和5AzA-cdR組ppENK的蛋白表達(dá)水平較對照組顯著升高,同時伴隨DNMT1和DNMT3a的蛋白表達(dá)減弱。
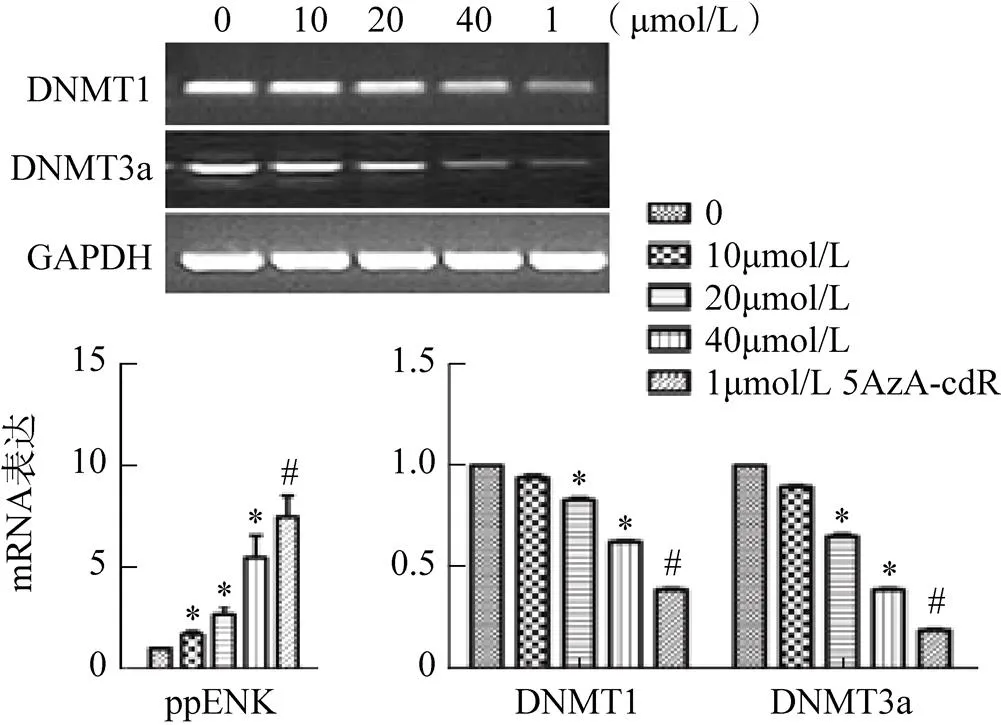
圖3 PCR實驗檢測結(jié)果
注:0為對照組,*<0.05,#<0.01
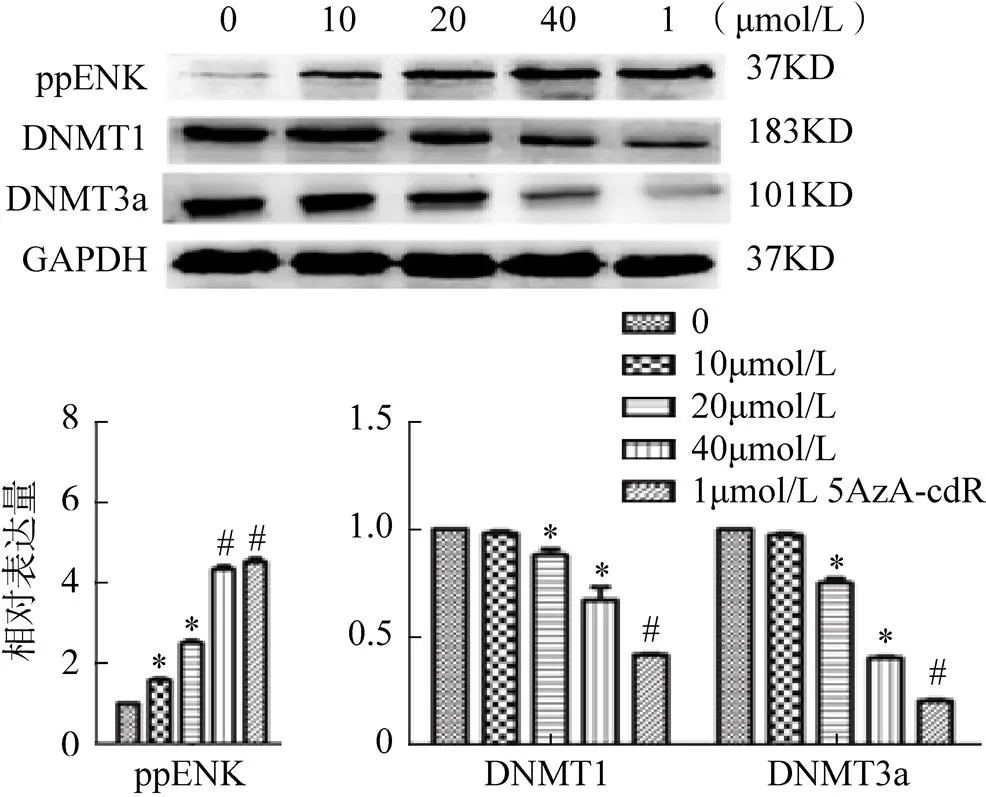
圖4 蛋白質(zhì)印跡(Western Blot,WB)實驗檢測結(jié)果
注:0為對照組;*<0.05,#<0.01
3 討論
胰腺癌發(fā)病率逐年升高,且病死率高。由于發(fā)病機(jī)制不明,胰腺癌的治療仍是一個重大挑戰(zhàn)[13]。大黃素對胰腺癌有一定的臨床療效,可誘導(dǎo)胰腺癌細(xì)胞凋亡,抑制新生血管形成,與吉西他濱合用可提高胰腺癌細(xì)胞對吉西他濱的耐藥性,但具體機(jī)制尚不清楚[12-14]。
胰腺癌抑癌基因的啟動子區(qū)CpG島甲基化被認(rèn)為是表達(dá)失活的主要原因之一[15]。ppENK基因與腫瘤細(xì)胞生長遲緩有關(guān),ppENK基因位于8q23~24,由4個外顯子和3個內(nèi)含子組成,ppENK基因編碼的產(chǎn)物是阿片類生長因子,對包括胰腺癌在內(nèi)的多種腫瘤都具有生長抑制作用,在胰腺癌細(xì)胞中檢測到ppENK的高甲基化[16]。本課題組既往MSP實驗只能定性檢測甲基化的狀態(tài),不能定量反應(yīng)甲基化的比例,故本研究應(yīng)用BSP實驗定量檢測甲基化水平,數(shù)據(jù)顯示在0μmol/L、10μmol/L、20μmol/L和40μmol/L的大黃素作用下,ppENK甲基化率分別為86.4%、82.7%、67.3%、52.7%,說明大黃素可對Panc1細(xì)胞的ppENK發(fā)揮不同程度的去甲基化作用。甲基化常導(dǎo)致抑癌基因表達(dá)缺失,其表達(dá)量與CpG島甲基化密度成反比,往往較低水平的甲基化導(dǎo)致抑癌基因失表達(dá),高密度的甲基化則可引起抑癌基因的完全失表達(dá)[17]。PCR結(jié)果也證實大黃素可使ppENK重新表達(dá),隨著藥物濃度的增加和作用時間的延長,其表達(dá)量有增加的趨勢,Western Blot結(jié)果也與PCR結(jié)果一致,進(jìn)一步證明大黃素對Panc1細(xì)胞具有去甲基化作用。
在生物體中,甲基化反應(yīng)主要由甲基轉(zhuǎn)移酶(DNMT1、DNMT3a、DNMT3b)催化,抑癌基因去甲基化的途徑主要有抑制甲基轉(zhuǎn)移酶活性和降低甲基轉(zhuǎn)移酶表達(dá)兩種方式。根據(jù)本研究中PCR和Western Blot的結(jié)果,40μmol/L大黃素和1μmol/L 5AzA-cdR可顯著降低DNMT1和DNMT3a的表達(dá),因此推測大黃素發(fā)揮去甲基化作用可能與其一定程度上抑制甲基轉(zhuǎn)移酶的表達(dá)有關(guān),能否抑制甲基轉(zhuǎn)移酶活性有待后續(xù)研究。
綜上所述,BSP證實大黃素可在一定程度上使Panc1中的抑癌基因ppENK去甲基化,PCR及Western Blot結(jié)果證明大黃素可增強(qiáng)ppENK的表達(dá),減弱DNMT1和DNMT3a的表達(dá),證明大黃素可通過抑制DNMTs的表達(dá)發(fā)揮其去甲基化的作用。本結(jié)果為大黃素治療胰腺癌的作用機(jī)制提供了新的認(rèn)識,在臨床上,大黃素治療胰腺癌不僅是通過抑制細(xì)胞凋亡和血管生成,表觀遺傳學(xué)中的去甲基化作用也是一個重要機(jī)制。
[1] SUNG H, FERLAY J, SIEGEL R L, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries[J]. CA Cancer J Clin, 2021, 71(3): 209–249.
[2] SIEGEL R L, MILLER K D, FUCHS H E, et al. Cancer statistics, 2021[J]. CA Cancer J Clin, 2021, 71(1): 7–33.
[3] RAHIB L, SMITH B D, AIZENBERG R, et al. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreascancers in the United States[J]. Cancer Res, 2014, 74(11): 2913–2921.
[4] CHENG X, BLUMENTHAL R M. Mammalian DNA methyltransferases: A structural perspective[J]. Structure, 2008, 16(3): 341–350.
[5] BLAZE J, WANG J, HO L, et al. Polyphenolic compounds alter stress-induced patterns of global DNA methylation in brain and blood[J]. Mol Nutr Food Res, 2018, 62(8): 1700722.
[6] MATSUOKA S, EDWARDS M C, BAI C, et al. P57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene[J]. Genes Dev, 1995, 9(6): 650–662.
[7] SATO N, FUKUSHIMA N, MAEHARA N, et al. SPARC/osteonectin is a frequent target for aberrant methylation in pancreatic adenocarcinoma and a mediator of tumor-stromal interactions[J]. Oncogene, 2003, 22(32): 5021–5030.
[8] ATTRI J, SRINIVASAN R, MAJUMDAR S, et al. Alterations of tumor suppressor gene p16INK4a in pancreatic ductal carcinoma[J]. BMC Gastroenterol, 2005, 5: 22.
[9] DAMMANN R, SCHAGDARSURENGIN U, LIU L, et al. Frequent RASSF1A promoter hypermethylation and K-ras mutations in pancreatic carcinoma[J]. Oncogene, 2003, 22(24): 3806–3812.
[10] UEKI T, TOYOTA M, SKINNER H, et al. Identification and characterization of differentially methylated CpG islands in pancreatic carcinoma[J]. Cancer Res, 2011, 61(23): 8540–8546.
[11] FUKUSHIMA N, SATO N, UEKI T, et al. Aberrant methylation of preproenkephalin and p16 genes in pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma[J]. Am J Pathol, 2002, 160(5): 1573–1581.
[12] SHENG Z L, WEI T W, HUI C, et al. Antitumor activity of emodin against pancreatic cancer depends on its dual role: Promotion of apoptosis and suppression of angiogenesis[J]. PLoS One, 2012, 7(8): e42146.
[13] LIU A, LUO J, ZHANG J H. Emodin combined gemcitabine inhibited the growth of pancreatic cancer in vitro and in vivo and its mechanisms study[J]. Zhongguo Zhong Xi Yi Jie He Za Zhi, 2012, 32(5): 652–656.
[14] CHEN H, WEI W, GUO Y, et al. Enhanced effect of gemcitabine by emodin against pancreatic cancer in vivo via cytochrome C-regulated apoptosis[J]. Oncol Rep, 2011, 25: 1253–1261.
[15] UEKI T, TOYOTA M, SOHN T, et al. Hypermethylation of multiple genes in pancreatic adenocarcinoma[J]. Cancer Res, 2000, 60(7): 1835–1839.
[16] DENNING G M, ACKERMANN L W, BARNA T J, et al. Proenkepha-lin expression and enkephalin release are widely observed in non-neuronal tissues[J]. Peptides, 2008, 29(1): 83–92.
[17] TAHILIANI M, KOH K P, SHEN Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1[J]. Science, 2009, 324: 930–935.
Emodin plays a demethylation role in tumor suppressor gene ppENK by reducing the expression of DNMTs in pancreatic cancer cells
Department of Head and Neck Surgery, Jiaxing First Hospital, Jiaxing 314000, Zhejiang, China
To study the effect of emodin on pancreatic cancer cell Panc1 DNA methyltransferases (DNMTs) and its demethylation of CpG island in the promoter region of tumor suppressor gene pre-proenkephalin (ppENK), and to explore whether emodin can reverse the methylation of ppENK by reducing the expression of DNMTs.CCK-8 was used to detect the effect of different concentrations of emodin on the growth of Panc1 cells, and to explore the optimal drug concentration. Bisulfite genomic sequencing PCR (BSP) was used to detect the effect of different concentrations of emodin on the methylation status of ppENK gene. PCR and Western Blot were used to detect the mRNA and protein expression of ppENK and methyltransferases DNMT1 and DNMT3a.Emodin inhibited the growth of Panc1 cells in a time-dependent and concentration-dependent manner, half maximal inhibitory concentration (IC50) was approximately 40μmol/L, emodin can weaken the methylated state of ppENK and enhance the non-methylated state. Emodin can weaken the mRNA and protein expression of DNMTs and enhance the mRNA and protein expression of ppENK.Emodin can demethylate the tumor suppressor gene of Panc1 ppENK in varying degrees and re-express the tumor suppressor gene. The inhibition of emodin on the growth of pancreatic cancer cells is related to its demethylation. It is speculated that the demethylation effect of emodin is related to its inhibition on the expression of methyltransferase.
Emodin; Pancreatic cancer; Demethylation; DNMTs; BSP
R733
A
10.3969/j.issn.1673-9701.2023.30.002
浙江省基礎(chǔ)公益研究計劃項目(LQ20H160059)
陳亮,電子信箱:jxsdyyy10711@163.com
(2023–03–02)
(2023–10–24)
