穩定性重氮色鹽對蠶絲的低溫偶合染色性能
2023-11-14 01:00:56李學崇陳維國崔志華宋秋亞李惠軍
絲綢 2023年11期
李學崇, 陳維國, 崔志華,, 郭 慶, 宋秋亞, 李惠軍
(1.浙江理工大學 紡織科學與工程學院,杭州 310018; 2.先進紡織材料與制備技術教育部重點實驗室,杭州 310018;3.國家先進印染技術創新中心,山東 泰安 271000; 4.杭州華絲夏莎紡織科技有限公司,杭州 310018)
中國是絲綢的原產地,具有悠久的絲綢生產歷史。絲綢紡織品具有手感柔軟、輕盈光滑、穿著舒適等優點,是一種奢華的服裝面料。絲素蛋白是蠶絲纖維的主要組成部分,占蠶絲總質量的75%左右[1-3]。絲素蛋白按聚集態結構分為結晶態和無定形態兩部分[4]。結晶區域的肽鏈主要由甘氨酸、丙氨酸、絲氨酸等按序排列,在這種規則的排列下具有穩定的性質[5-6]。無定形區主要由色氨酸、酪氨酸和組氨酸等組成[7],該區域結構疏松,具有許多含活潑官能團的氨基酸殘基,是蠶絲與染料及其他物質發生化學反應的主要區域[8-9]。
目前蠶絲染色常用的酸性染料和絲素大分子之間以離子鍵、范德華力和氫鍵結合,耐濕處理牢度不理想[10];活性染料雖能與蠶絲反應生成共價鍵結合,但在染色過程中需高溫上染、高溫堿固色,對纖維的結構和物理機械性能帶來不利影響[11]。同時,酸性染料和活性染料均需要用大量的鹽來促染,造成了污水處理及環境的嚴重負擔。
不溶性偶氮染料用于棉纖維染色具有很長的歷史,雖然已被活性染料所替代,但這是一種低溫染色方法。色基在冰浴中重氮化后,在室溫狀態下和預先處理到織物上的色酚發生偶合反應,得到給色量高、色澤鮮艷的染色印花產品。蠶絲中酪氨酸殘基占其總量的6.4%[12],主要位于無定形區,酪氨酸殘基側基是活性染料反應的位點[13],而酚羥基的供電效應使其鄰位碳易發生親電取代反應,也為重氮鹽與酪氨酸殘基發生偶合反應提供了反應位點[14],從而使得蠶絲能夠在不添加其他偶合組分的情況下實現與色基重氮鹽的共價鍵合反應,如圖1所示。
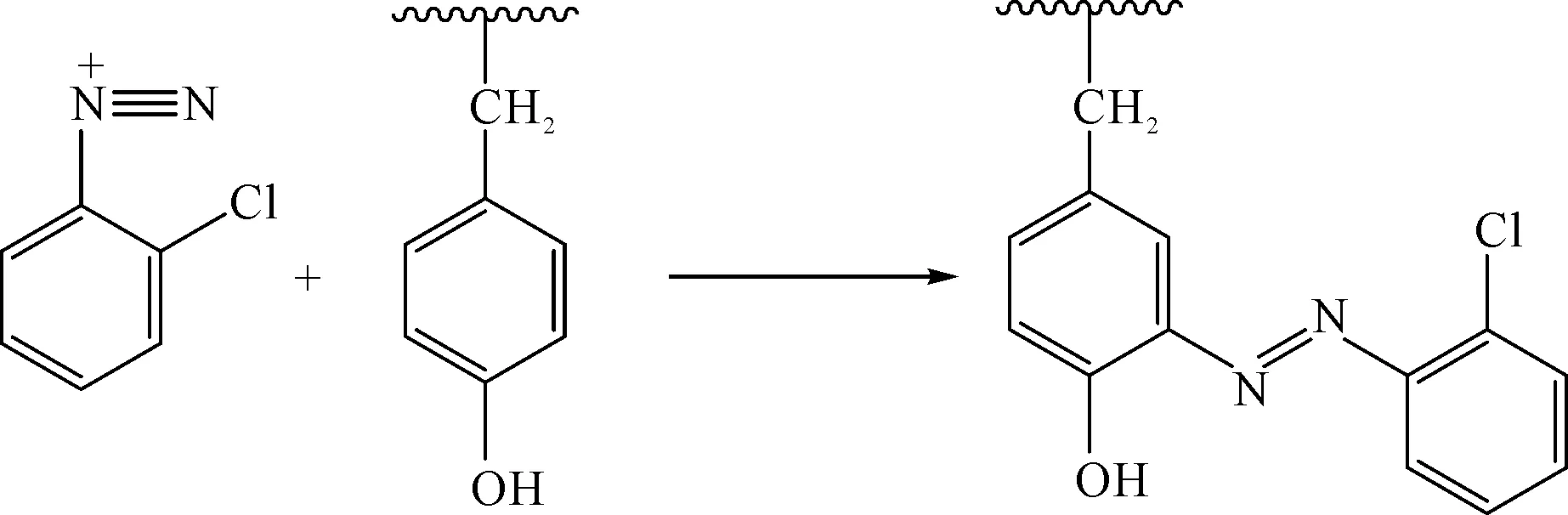
圖1 色基重氮鹽與絲素中酪氨酸殘基的偶合反應
現有研究顯示,Chen等[15]實現了芳胺衍生物制備的重氮鹽與蠶絲原位偶合,開發了一種耐濕處理且色牢度較好的絲綢染色方法。同時Chen等[16]以大紅色基G制備重氮鹽后與對甲基苯酚偶合,通過產物表征證明了絲綢和棉在傳統偶合顯色方法下的不同結果是由于在染色絲綢上形成了兩種發色團。Wang等[17]用1-氨基蒽醌重氮鹽與蠶絲偶聯染色,證明了常用蒽醌結構染料通過制備相應重氮鹽可以用于絲綢染色,所染絲綢表現出良好的色牢度和強度。江華等[18]揭示了具有供吸電性基團、大共軛體系或磺酸基團的芳胺重氮鹽結構對蠶絲偶合染色的影響規律,并優化了同浴加熱階段上染溫度、染浴pH值和元明粉質量濃度等染色條件,探討了該方法的染料使用范圍。
將芳胺重氮鹽直接應用于蠶絲偶合顯色,受到其穩定性等因素的限制。由于芳胺重氮鹽的耐熱穩定性較差,之前研究的芳胺重氮化及其與蠶絲的偶合過程都在低溫0~5 ℃下進行,需要消耗一定量的冰塊來降低溫度;同時為防止重氮鹽分解導致的物料損失,芳胺重氮化需要在偶合顯色前現場配制,操作不便。固體重氮色鹽具有一定的穩定性,可以在常溫下儲存一定時間,有利于工業化應用。
本文在有機溶劑二氯甲烷中制備四氟硼酸重氮鹽,在室溫條件下用四氟硼酸重氮色鹽對蠶絲進行偶合顯色,并對工藝進行優化,然后與冰浴重氮鹽溶液直接偶合染色進行對比,同時檢測不同存放時間后四氟硼酸重氮色鹽水溶液的吸光度以表征其儲存穩定性。將四氟硼酸重氮色鹽對蠶絲和棉分別進行偶合顯色反應,并對比其顯色結果以揭示穩定色鹽對蠶絲染色的反應機理。
1 材料與方法
1.1 實驗材料與儀器
材料:電力紡練白坯為平方米質量60 g/m2的蠶絲織物(杭州喜得寶集團有限公司),鹽酸、亞硝酸鈉、尿素、碳酸鈉、十二烷基苯磺酸鈉、無水乙醚、檸檬酸、磷酸氫二鈉等均為化學純(杭州高晶精細化工有限公司),鄰氯苯胺、2,5-二氯苯胺、三氟化硼乙醚、亞硝酸叔丁酯、二氯甲烷、正戊烷、色酚AS均為化學純(上海阿拉丁生化科技股份有限公司),滲透劑JFC為脂肪醇聚氧乙烯醚工業品(山東萬禾化工有限公司),H酸(1-氨基-8-萘酚-3,6-二磺酸)為工業品(江蘇從眾化工股份有限公司)。本實驗用于制備四氟硼酸重氮色鹽的兩個芳胺及其對應重氮化產物的化學結構如圖2所示。

圖2 鄰氯苯胺和2,5-二氯苯胺的重氮化
儀器:DF-101S集熱式恒溫加熱磁力攪拌器、PHS-2F型pH值計(上海儀電科學儀器股份有限公司),FD-1A-50冷凍干燥機(上海比朗儀器制造有限公司),LCQ-Fleet型質譜儀(美國Themo公司),AVANCE AV400MHz型核磁共振波譜儀(瑞士布魯克公司),NlcoletiS50型傅里葉變換紅外光譜儀(美國賽默飛公司),UV-2600型紫外分光光度儀(島津(上海)實驗器材有限公司),AS-12/24常溫振蕩式小樣機(佛山市精柯紡織印染設備有限公司),SF600X-PLUS型測色配色儀(美國Datacolor公司)。
1.2 四氟硼酸重氮色鹽的制備
參考Doyle等[19]四氟硼酸重氮色鹽的制備方法:將7.5 mmol的三氟化硼乙醚溶解于10 mL二氯甲烷加入三口燒瓶中并攪拌,整個裝置在冰水浴中冷卻至0~5 ℃。然后加入5 mmol芳胺攪拌至均勻,若形成了固體胺-BF3絡合物,則繼續加入二氯甲烷以產生均勻的溶液。在10 min內將5 mmol亞硝酸叔丁酯溶于5 mL二氯甲烷的溶液逐滴加入快速攪拌的反應溶液中。完全加入后,保持低溫(0~5 ℃)反應30 min,用薄層色譜跟蹤反應終點,展開劑為二氯甲烷-乙酸乙酯(體積比15︰3)混合溶劑。反應結束后向反應溶液中加入40 mL正戊烷,抽濾固體,以冷乙醚多次洗滌,濾餅置于冷凍干燥機中干燥8 h,得到純凈的四氟硼酸重氮鹽后置于裝有變色硅膠的離心管中,保存待用。
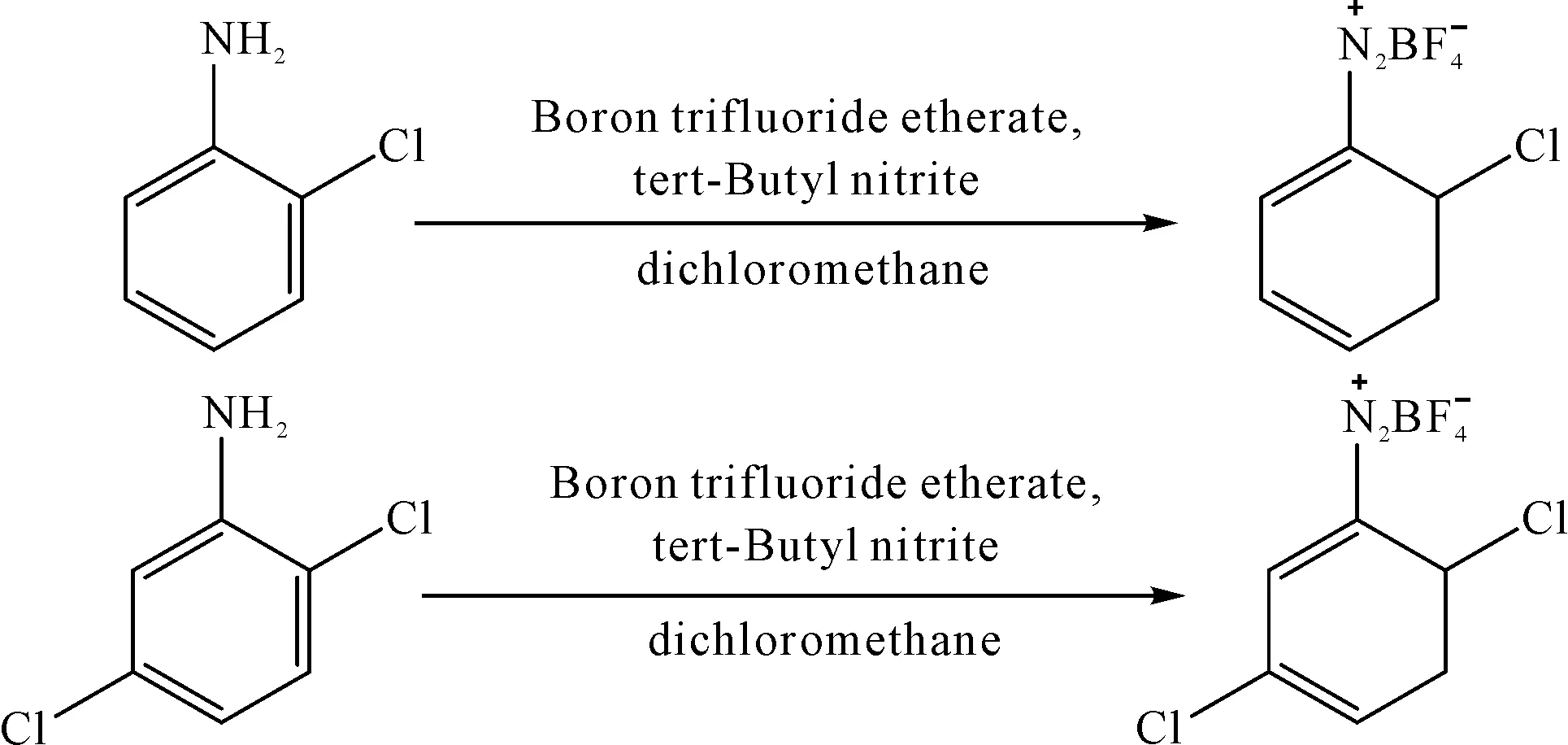
圖3 鄰氯苯胺及2,5-二氯苯胺的重氮色鹽制備
1.3 織物預處理工藝
1.3.1 蠶絲原位偶合預處理
常溫下,配制50 mL NaOH(1 g/L)水溶液,向溶液中加入10 g/L滲透劑JFC 1 mL。將1 g蠶絲(或棉)織物浸入溶液中快速潤濕,取出后軋去多余溶液,連續浸軋兩次,織物帶液率為100%。
1.3.2 蠶絲色酚偶合預處理
常溫下,將色酚AS(1 g/L)與乙醇攪拌成漿狀,攪拌下加入50 mL NaOH(1 g/L)水溶液,直至形成透明溶液,再向溶液中加入10 g/L滲透劑JFC 1 mL。將1 g蠶絲(或棉)織物浸入溶液中快速潤濕,取出后軋去多余溶液,連續浸軋兩次,織物帶液率為100%。
1.4 芳胺重氮液對蠶絲著色工藝
將0.5 mmol芳胺、0.1 mL濃鹽酸、10 mL去離子水、1 mL質量濃度為10 g/L的滲透劑JFC依次加入三口燒瓶中,攪拌至芳胺溶解。另取0.6 mmol亞硝酸鈉溶解于2 mL去離子水中,隨后在2~10 min內向三口燒瓶中逐滴加入亞硝酸鈉溶液并保持在0~5 ℃反應30 min,用淀粉-碘化鉀試紙檢測亞硝酸是否過量,用尿素除去過量亞硝酸。反應完成后將重氮鹽溶液加入到20 mL磷酸氫二鈉-檸檬酸緩沖溶液(pH值4.5)中并保持溶液環境溫度為0~5 ℃,將1 g經過預處理工藝(工藝條件見1.3)的蠶絲織物浸入上述混合溶液中,室溫浸染30 min后取出,用冷水充分洗滌后皂洗(皂洗工藝為皂片1 g/L,純堿1 g/L,溫度80 ℃,時間10 min,浴比50︰1),冷水洗滌,晾干。
將蠶絲或棉織物用色酚AS預處理后,與色鹽的顯色反應如圖4所示。
1.5 芳胺重氮色鹽對蠶絲及棉著色工藝
常溫下,將0.5 mmol色鹽、10 g/L滲透劑JFC 1 mL和10 mL去離子水混合于錐形瓶中,溶解后向溶液中加入20 mL磷酸氫二鈉-檸檬酸緩沖溶液(pH值4.5)。將1 g經過預處理工藝(工藝條件見1.3)的蠶絲織物(或棉織物)浸入上述混合溶液中,室溫浸染30 min后取出,用冷水充分洗滌后皂洗(皂洗工藝同1.4),冷水洗滌,晾干。
1.6 芳胺重氮液及色鹽的存儲穩定性
將鄰氯苯胺按照1.2方法制備色鹽,按照1.4方法制備其重氮化后溶液。
另配制1 mol/L H酸溶液50 mL,加入20 mL磷酸氫二鈉-檸檬酸緩沖溶液(pH值8)。芳胺重氮鹽與H酸顯色反應如圖5所示。

圖4 鄰氯苯重氮色鹽與色酚AS偶合
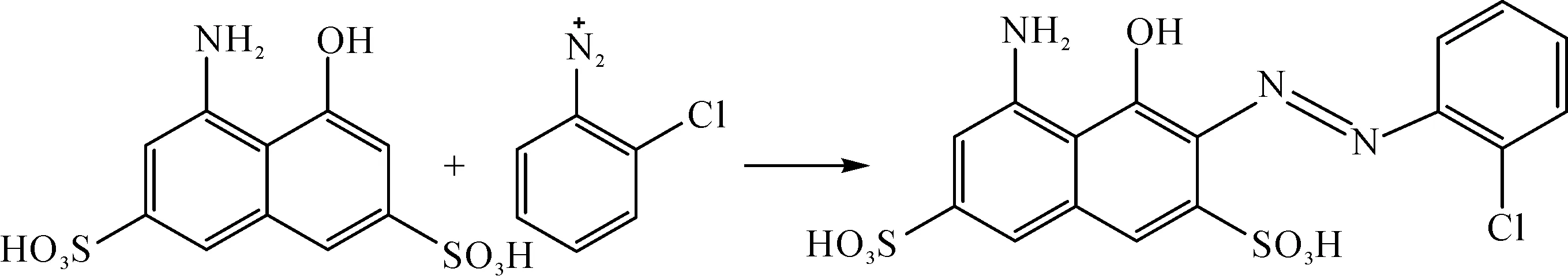
圖5 H酸與芳胺重氮鹽的偶合顯色反應
為了考察鄰氯苯胺重氮化溶液的存儲穩定性,本文將鄰氯苯胺按照1.4方法制備其對應的重氮化溶液,將其與過量的偶合組分H酸發生反應生成偶氮發色體顯色液,取顯色液并對其進行紫外-可見光吸收光譜測試。
1) 取剛制備的重氮液1.2 mL加入2.0 mL H酸溶液中,充分反應,獲得顯色液A。2) 上述重氮液常溫存放3 d,再取1.2 mL重氮液與2.0 mL H酸溶液反應獲得顯色液B。3) 取剛制備的色鹽0.05 mmol溶于1.2 mL水中,與2.0 mL H酸溶液反應獲得顯色液C。4) 取剛制備的色鹽0.05 mmol溶于1.2 mL水中,常溫存放3 d后與2 mL H酸溶液反應獲得顯色液D。5) 制備的色鹽以固體形式在常溫條件下保存3 d后,取0.05 mmol溶于1.2 mL水中,與2.0 mL H酸溶液反應獲得顯色液E。對顯色液A、B、C、D、E分別進行紫外-可見光吸收光譜測試。
為了評價直接制備的芳胺重氮液和芳胺重氮色鹽的存儲穩定性,本文定義其水溶液的吸光度變化率,并按照下式計算:
L/%=(1-A1/A0)×100
(1)
式中:L為吸光度變化率;A1為某時刻最大吸收波長處的吸光度;A0為存儲開始時刻最大吸收波長處的吸光度。
1.7 織物染色性能測試
1.7.1 色深值K/S值測試
染色織物折疊至沒有明顯透光,使用測色配色儀測試其3個隨機位點的K/S值,取平均值。
1.7.2 色牢度測試
染色織物耐皂洗色牢度按照GB/T 3921—2008《紡織品 色牢度試驗耐皂洗色牢度》進行測試;染色織物耐摩擦色牢度按照GB/T 3920—2008《紡織品 色牢度試驗耐摩擦色牢度》進行測試。
2 結果與分析
2.1 四氟硼酸重氮色鹽結構表征
2.1.1 傅里葉紅外光譜分析
將制備的鄰氯苯重氮色鹽進行紅外光譜測試,結果如圖6所示。
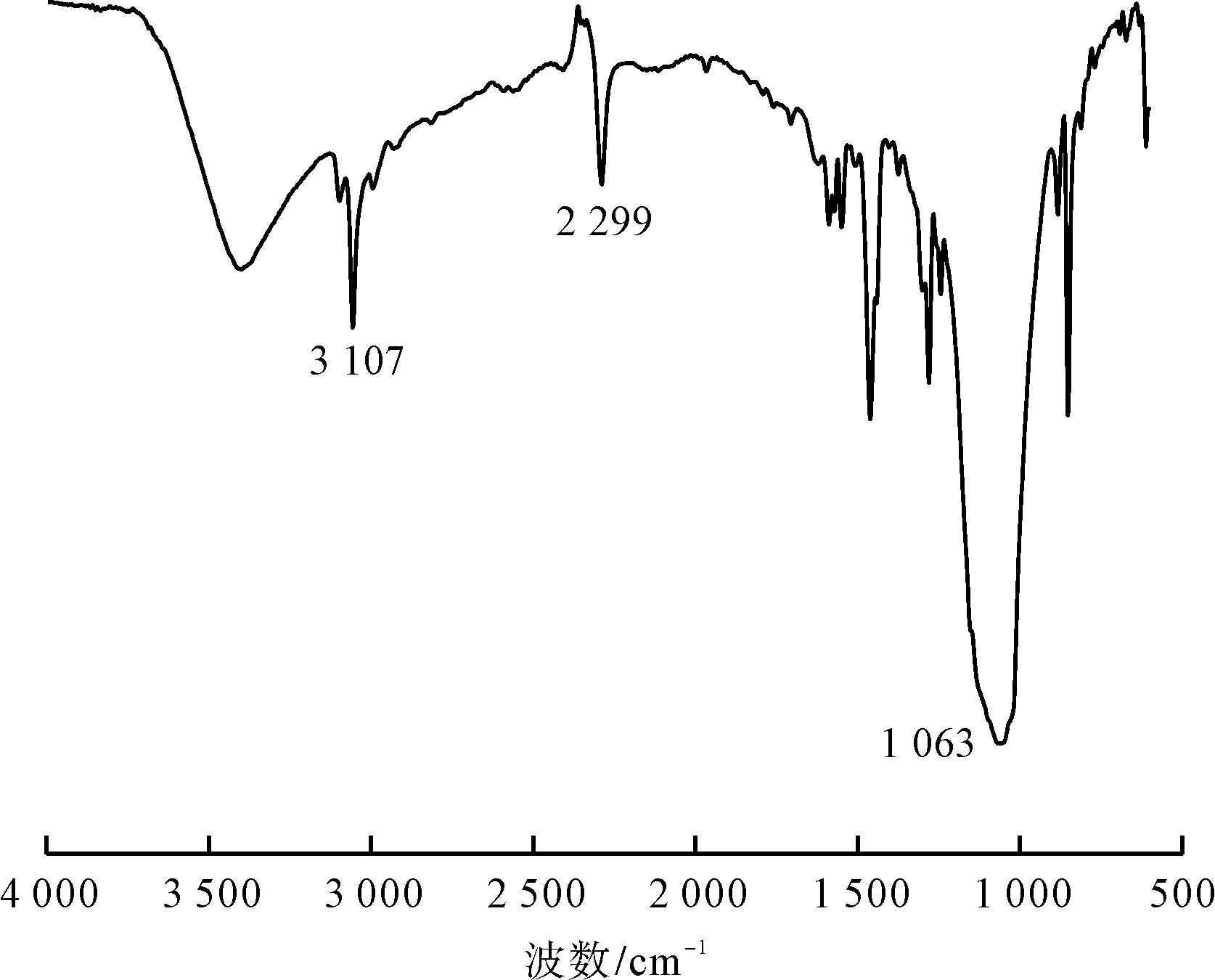
圖6 鄰氯苯重氮色鹽的紅外光譜圖
由圖6可知,3 107 cm-1為分子結構中苯環上C—H伸縮振動吸收峰,2 299 cm-1為分子結構中—N+≡N伸縮振動吸收峰,1 063 cm-1為分子結構中苯環上C—Cl伸縮振動吸收峰。通過上述基團特征吸收峰分析,佐證了合成目標鄰氯苯重氮色鹽的分子結構。
將2,5-二氯苯重氮色鹽進行紅外光譜測試,結果如圖7所示。
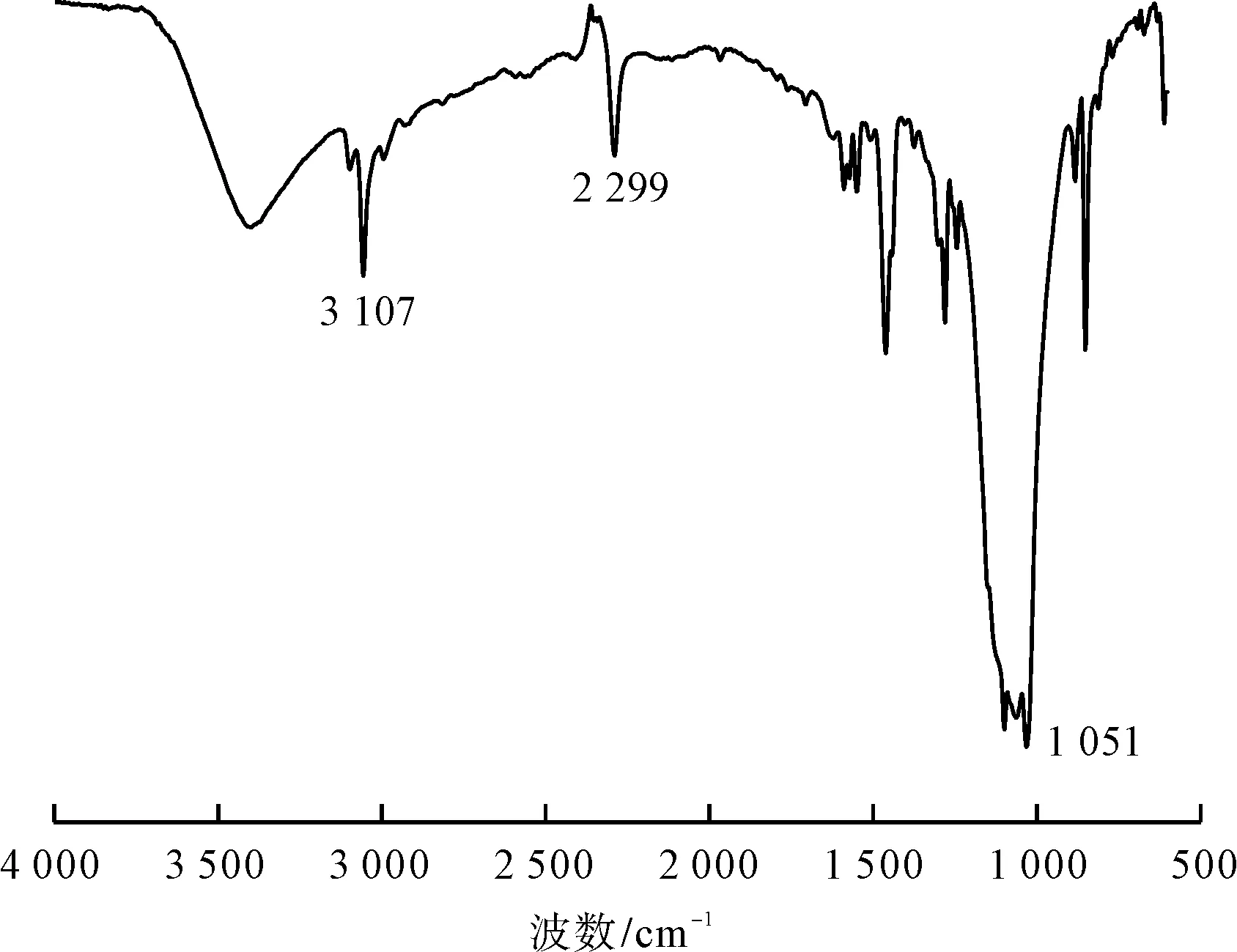
圖7 2,5-二氯苯重氮色鹽的紅外光譜圖
由圖7可知,3 107 cm-1為分子結構中苯環上C—H伸縮振動吸收峰,2 299 cm-1為分子結構中氮氮三鍵伸縮振動吸收峰,1 051 cm-1為分子結構中苯環上C—Cl伸縮振動吸收峰。通過上述基團特征吸收峰分析,佐證了合成目標2,5-二氯苯重氮色鹽的分子結構。
2.1.2 核磁共振氫譜分析
鄰氯苯重氮色鹽的核磁共振氫譜如圖8所示。
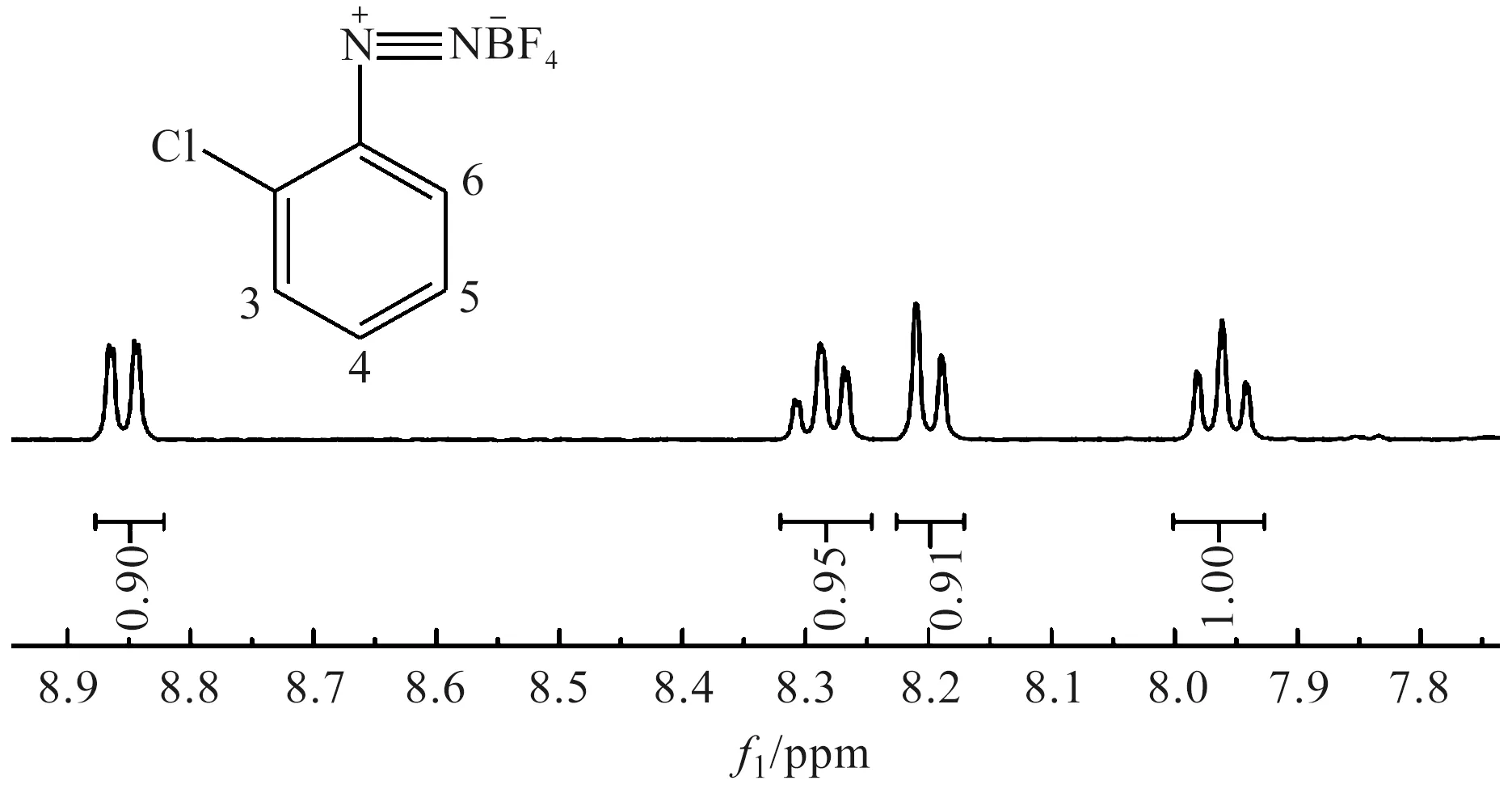
圖8 鄰氯苯重氮色鹽的核磁共振氫譜圖
鄰氯苯重氮色鹽的1H NMR解析:1H NMR(400 MHz,DMSO-d6)δ8.85(d,J=8.3 Hz,1H),8.28(t,J=8.0 Hz,1H),8.20(d,J=8.3 Hz,1H),7.96(t,J=8.0 Hz,1H)。
2,5-二氯苯重氮色鹽的核磁共振氫譜如圖9所示。
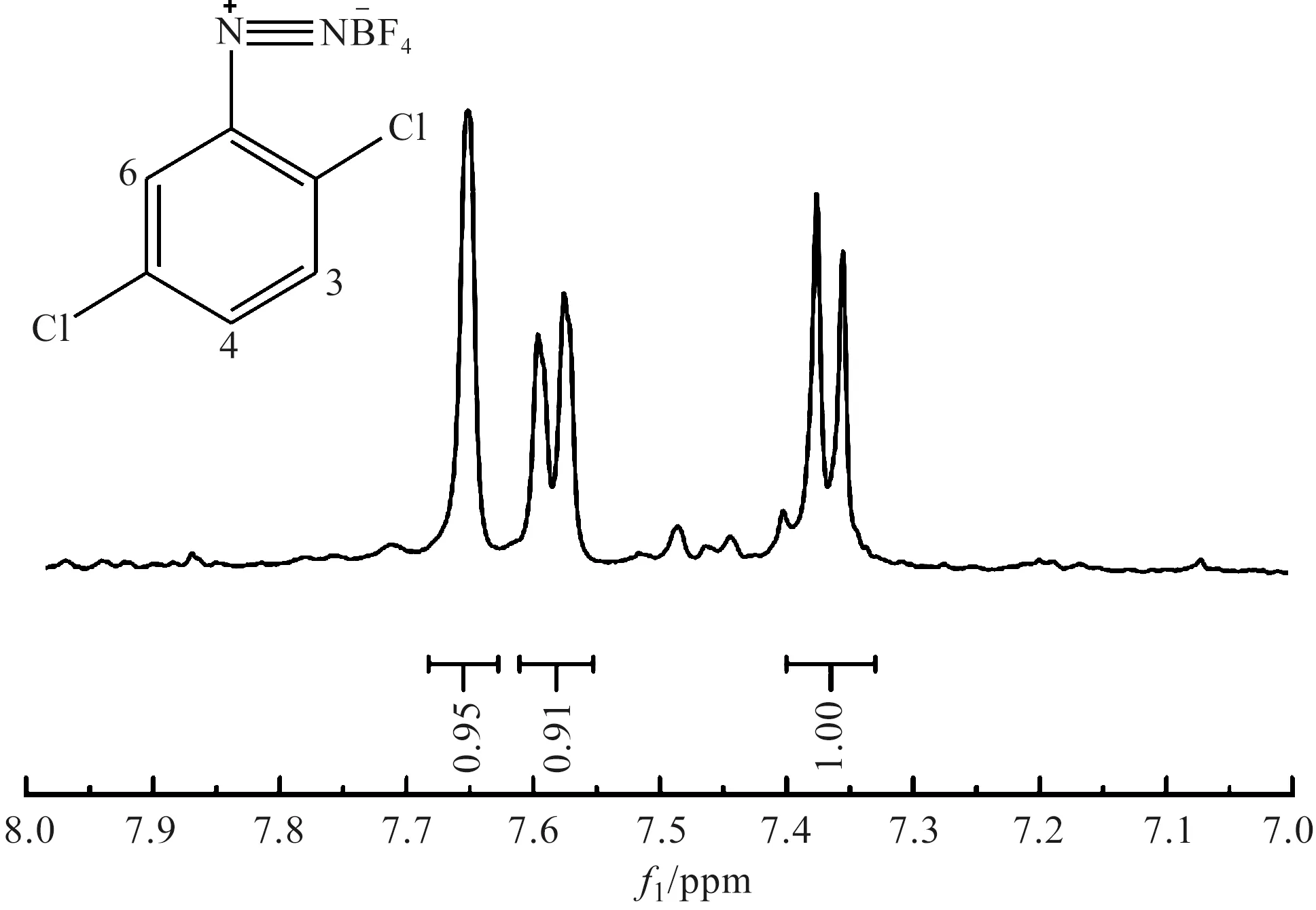
圖9 2,5-二氯苯重氮色鹽的核磁共振氫譜圖
2,5-二氯苯重氮色鹽的1H NMR解析:1H NMR(400 MHz,DMSO-d6)δ7.65(d,J=2.1 Hz,1H),7.58(t,J=2.1 Hz,1H),7.37(d,J=8.2 Hz,1H)。
綜上,鄰氯苯重氮色鹽的產率為72.24%;2,5-二氯苯重氮色鹽的產率為70.89%。
2.2 色鹽的存儲穩定性
按1.6方法生成偶氮發色體顯色液A與顯色液B,顯色液A及顯色液B的測試結果如圖10(a)所示。顯然,重氮液制備后馬上與H酸反應形成了對應顏色的產物(圖5),顯色液A的紫外—可見吸收光譜圖中,在491 nm處有吸光度為1.3的最大吸收峰;顯色液B的測試結果表明,當芳胺重氮鹽溶液在常溫存放3 d后,僅存在微量未分解重氮鹽與H酸進行顯色反應,也即芳胺重氮鹽大部分已發生分解。顯然,芳胺重氮鹽溶液的儲存穩定性很差,制備后要馬上使用。
按1.2方法將制備好的鄰氯苯重氮色鹽溶于水,將其與過量H酸溶液反應后進行紫外-可見光吸收光譜測試。按照1.6方法,得到剛制備的色鹽與H酸反應的顯色液C、制備后以溶液形式存放3 d后與H酸反應的顯色液D,以及制備后以固體形式存放3 d后與H酸反應的顯色液E,分別對顯色液C、顯色液D、顯色液E進行紫外-可見光吸收光譜測試,測試結果如圖10(b)所示。在圖10(b)中,重氮化色鹽以固體形式存放3 d后的顯色液的紫外-可見光吸收光譜變化不大,而以溶液形式存放3 d后的顯色液吸光譜變化較為明顯。如圖10(a)所示,按1.6中L(吸光度變化率)的計算公式得到重氮液制備后存放3 d后的吸光度變化率為94.5%(顯色液B與顯色液A對比);如圖10(b)所示,色鹽制備后以溶液形式存放3 d后的吸光度變化率為20.7%(顯色液D與顯色液C對比),色鹽制備后以固體形式存放3 d后的吸光度變化率為3.5%(顯色液E與顯色液C對比)。由此可知,四氟硼酸重氮色鹽具有相當好的存儲穩定性,以固體形式存放比溶液形式存放更有利于色鹽有效成分的保存。
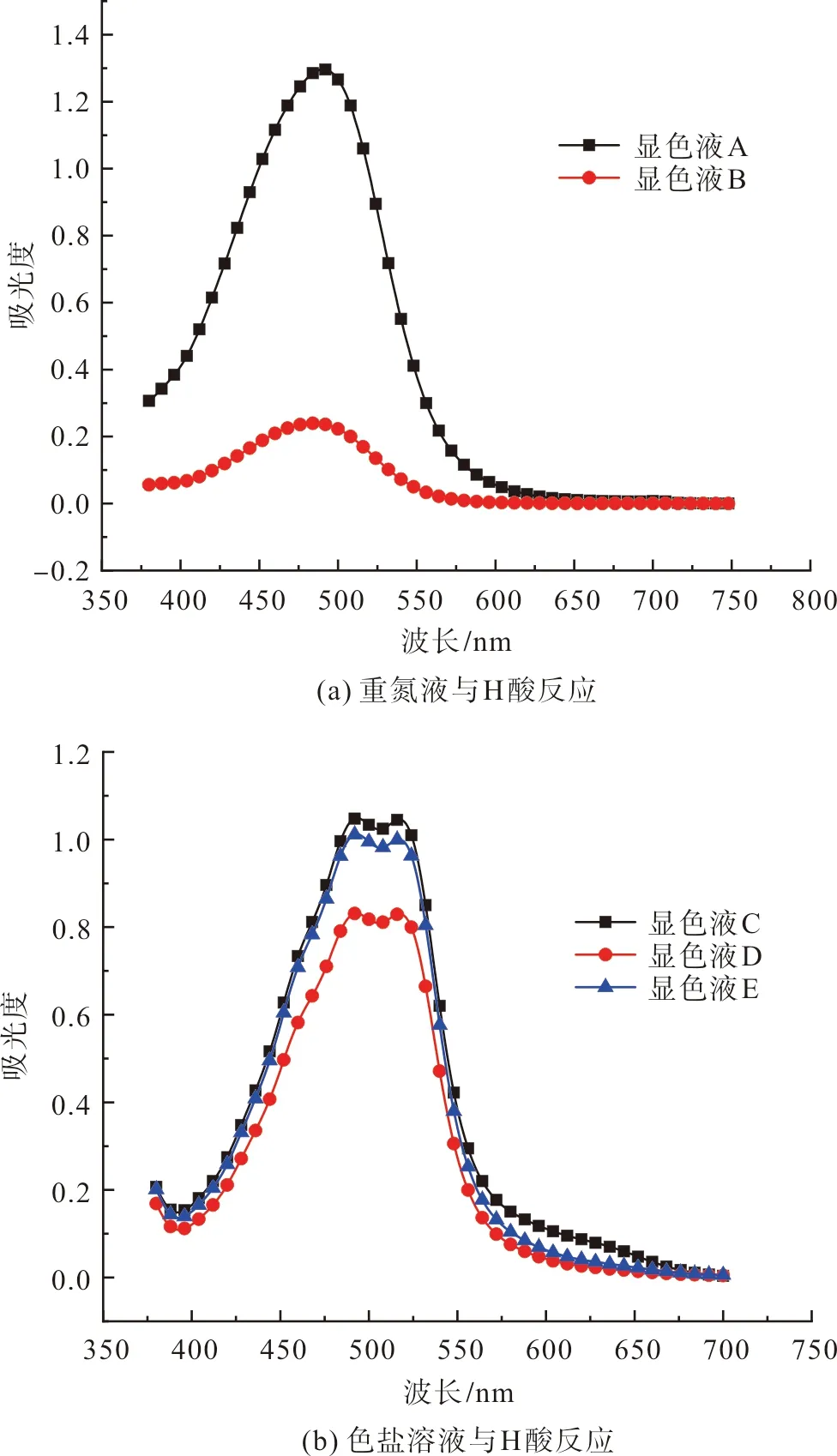
圖10 偶氮發色體顯色液的紫外-可見光吸收光譜
2.3 蠶絲織物的偶合顯色
在成功制備色鹽并驗證其存儲穩定性后,將兩種色鹽應用于蠶絲染色,顯色結果如圖11所示。
圖11(a)中,兩條K/S值曲線分別表示的是鄰氯苯重氮色鹽與蠶絲原位偶合、色鹽與浸軋色酚AS前處理的蠶絲偶合。其中,蠶絲原位偶合染色的蠶絲織物為黃色(λmax=400 nm;L*a*b:76.6,27.2,86.4),浸軋色酚AS后的蠶絲織物偶合染色結果為橙紅色(λmax=500 nm;L*a*b:57.8,67.3,63.9)。
圖11(b)中,兩條K/S值曲線分別表示的是2,5-二氯苯重氮色鹽與蠶絲原位偶合、色鹽與浸軋色酚AS前處理的蠶絲偶合。其中,原位偶合的蠶絲織物為黃色(λmax=440 nm;L*a*b:65.6,35.4,75.1),浸軋色酚AS后的蠶絲織物偶合染色結果為橙紅色(λmax=440 nm;L*a*b:58.1,65.1,64.6)。

圖11 重氮色鹽與蠶絲的偶合顯色(單獨或色酚AS存在下)
2.4 色基重氮鹽與色鹽的染色性差異分析
將色基重氮鹽溶液直接用于偶合顯色(按照1.4)與固體重氮色鹽溶液用于偶合顯色(按照1.5)作對比。
由于重氮液中含有尿素、NaCl,成分復雜,同時固體四氟硼酸重氮鹽中可能含有雜質,為了排除這些因素對最后染色效果的影響,需要對用量進行統一。故本文以已知摩爾濃度的鄰氯苯胺色基重氮液的最大吸收波長處吸光度,在色鹽溶液標準工作曲線上標定對應的色鹽摩爾濃度,如圖12所示。

圖12 色鹽溶液的標準工作曲線
鄰氯苯重氮色鹽溶液的最大吸收波長為414 nm。由圖12可見,當色基重氮液摩爾濃度為15 mmol/L,對應的重氮色鹽溶液摩爾濃度為15.37 mmol/L。故后續工藝按上述用量進行。
圖13為兩種芳胺重氮液與色鹽分別原位染色蠶絲及加入色酚AS后偶合染色蠶絲的結果,通過對比圖13中的重氮液及重氮色鹽染色蠶絲后的K/S值曲線,無論有無色酚AS的加入,重氮液與重氮色鹽的染色效果都近似。而圖14中,色鹽對棉染色的K/S值幾乎為零,說明色鹽與棉纖維不發生偶合反應,沒有在纖維上合成發色體,當棉織物預處理上色酚AS后,能夠與色鹽發生反應并顯色。這是由于色鹽與色酚AS發生了重氮偶合反應,生成了發色體,能夠證明該實驗條件下唯一的發色體為色鹽與色酚AS的偶合產物。再通過對比圖14中棉織物與圖13中蠶絲織物的K/S值曲線,最大吸收波長的變化是由于色鹽與蠶絲酪氨酸殘基及色酚AS均發生了偶合反應,兩種發色體產生了復合顯色作用。

圖13 芳胺重氮液或重氮色鹽對蠶絲(單獨或色酚AS存在下)偶合染色
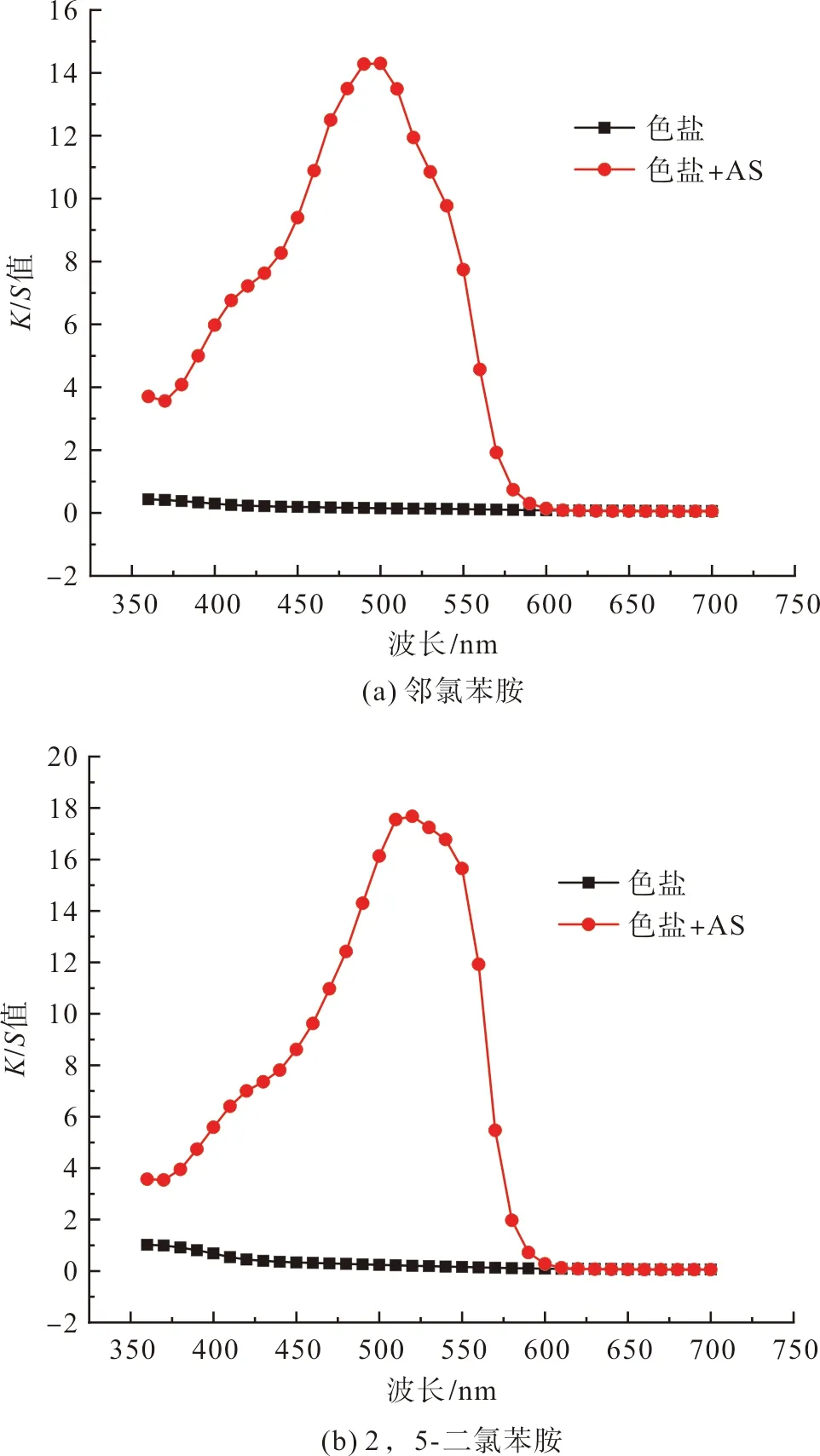
圖14 重氮色鹽對棉(單獨或色酚AS存在下)偶合染色
2.5 四氟硼酸重氮色鹽對蠶絲織物偶合染色的色牢度
采用兩種芳胺制備對應的四氟硼酸重氮色鹽后對蠶絲進行偶合染色,其耐皂洗色牢度和耐摩擦色牢度如表1所示。由表1可知,經上述方法染色后的蠶絲織物耐皂洗色牢度和耐摩擦色牢度均在4級以上。依據GB/T 15551—2016《桑蠶絲織物》中桑蠶絲織物內在質量分等規定,重氮色鹽偶合染色蠶絲織物的耐皂洗色牢度、耐摩擦色牢度均已達到優等品的要求。

表1 四氟硼酸重氮色鹽對蠶絲織物偶合染色的色牢度
3 結 論
本文針對蠶絲染色中存在的常用酸性染料濕處理色牢度低、活性染料易水解及大量用鹽等問題,開展了穩定性重氮色鹽制備及其對蠶絲低溫偶合染色性能的研究。得到結論如下:
1) 成功制備了鄰氯苯胺和2,5-二氯苯胺的四氟硼酸重氮色鹽,其中吸電子基團的引入有助于進一步提高四氟硼酸重氮鹽的存儲穩定性。
2) 采用色鹽溶液與酪氨酸殘基原位偶合,所染蠶絲織物的耐皂洗色牢度和耐摩擦色牢度均在4級以上,滿足蠶絲織物高色牢度染色的要求。
3) 將穩定性色鹽低溫原位偶合與色酚偶合相結合,可以在蠶絲織物上獲得兩者的復配顏色,豐富了穩定性色鹽低溫偶合染色蠶絲織物的色彩。

《絲綢》官網下載

中國知網下載
