高壓下ZrN2晶體的結構、力學和電子性質的研究
2023-11-20 04:06:38戴偉邵小倩馮全超陳艷肖循蔣再富
長江大學學報(自科版) 2023年6期
戴偉,邵小倩,馮全超,陳艷,肖循,蔣再富
1.長江大學物理與光電工程學院,湖北 荊州 434023
2.荊楚理工學院數理學院,湖北 荊門 448000
過渡金屬氮化物具有高硬度、高熔點、高耐磨性和優越的電學性能,使其在切割和拋光工具,涂層材料,磨料等工業領域具有廣泛的應用前景[1-3]。氮化鋯作為過渡金屬氮化物的一種,因其良好的力學和光學性質,引起了人們的廣泛的關注[4-5]。GRIBAUDO等[6]通過研究Zr-N體系相圖,發現常溫下ZrN和Zr3N4是穩定的。ZERR等[7]在16 GPa和2 500 K的條件下合成了具有Th3P4結構的c-Zr3N4晶體,其維氏硬度約30 GPa。HAO等[8]研究了ZrN的結構相變和彈性性質,發現在約210.41 GPa的條件下發生B1→B2的結構相變。CHEN等[9]計算的ZrN的超導轉變溫度為10 K,可作為潛在的超導材料。WEN等[10]報道了Zr2N在0~200 GPa的壓力范圍內將經過P42/mnm→Pnnm→Cmcm→P4/nmm→I4/mcm的結構相變。YU等[11]采用第一性原理研究了Zr-N晶體的相變和力學性質,研究表明,低溫下ZrN、Zr2N、Zr4N3、Zr6N5、Zr8N7、Zr15N16、Zr7N8和Zr4N5具有熱力學穩定,Zr3N2在高溫下具有穩定性。盡管人們對Zr-N體系的晶體結構和性質進行了一些報道,但是,關于ZrN2化合物在高壓下的晶體結構、力學及電子性質的研究鮮有報道。
因此,本文使用CALYPSO程序包[12]在0~100 GPa壓力范圍內,對Zr-N體系在高壓下的穩定或亞穩態結構進行了搜索。基于密度泛函理論的第一性原理,計算ZrN2化合物的高壓相圖、聲子譜和彈性常數,進而探究其在高壓下的結構演化、穩定性及力學性能。此外,通過對ZrN2化合物的能帶結構和態密度計算,分析了它們的電子性質。
1 計算方法
采用基于粒子群優化方法的CALYPSO程序對ZrN2化合物在高壓下的結構進行搜索[12],利用基于密度泛函理論(DFT)的VASP軟件進行結構優化、力學和電子性質計算[13]。計算中,交換關聯泛函采用廣義梯度近似(GGA)中的Perdew-Burke-Ernzerhof(PBE)函數[14];使用投影增強波(PAW)方法描述Zr-N體系中的電子和離子間的相互作用[15]。根據收斂測試結果,將截止能量設置為600 eV,選擇稠密的Monkhorst-Pack k網格[16],以確保所有的焓值計算都收斂于1 meV/atom。聲子譜的計算采用直接超胞方法結合線性響應理論在PHONOPY程序中實現[17]。
2 結果與討論
2.1 結構預測及高壓下的相變
穩定的Zr-N化學計量晶體結構已通過凸包圖確定,其中熱力學穩定的化合物落在直線上。高壓下Zr-N體系在0 K時的穩定性可以根據ZrmNn晶體的形成焓來判斷,其計算公式如下:
ΔH=[H(ZrmNn)-mH(Zr)-nH(N2)/2]/(m+n)
(1)
式中:ΔH為氮化鋯晶體的形成焓;H(ZrmNn)為該化合物的每個晶胞的焓值;H(Zr)和H(N2)分別為每個Zr原子和N2分子處于基態時對應的焓值。
Zr-N體系相對Zr和N2的不同化學計量的能量穩定性反映在凸包圖中,如圖1所示。熱力學穩定結構用黑色實心方塊表示,并用黑色實線連接,紅色虛線連接代表亞穩態的紅色實心圓圈。在0 GPa時,沒有熱力學穩定的ZrN2化合物落在凸包上,僅有單斜相C2/m-ZrN2作為熱力學亞穩態結構出現。當壓力增加到50和100 GPa時,正交相Pnma-ZrN2恰好位于凸包上,是具有熱力學穩定性的結構。本文將重點研究0 GPa時C2/m-ZrN2相和50 GPa時Pnma-ZrN2相的結構特征和相關性質。
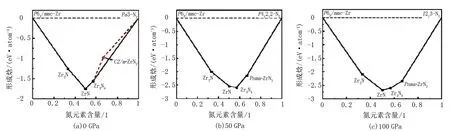
圖1 ZrN2化合物在不同壓力下的凸包圖
壓力對材料的結構影響很大,不同壓力下可能會發現不同的穩定結構。基于CALYPSO結構預測方法,對Zr-N二元體系進行了結構預測,確定了2個低能候選相,即單斜相C2/m-ZrN2和正交相Pnma-ZrN2。在特定壓力范圍內成功復現了已報道過的Zr2N,ZrN和Zr3N4結構,說明該方法適用于Zr-N體系。為了闡明ZrN2化合物在高壓下的相圖以及找到ZrN2化合物與其他氮化鋯之間的相對穩定性,構建了在0~100 GPa范圍內ZrN2候選結構相對于C2/m-ZrN2相的焓差與壓力的函數圖,如圖2(a)示。由圖2(a)可知,在低壓下,競爭相1/3(Zr3N4+N2)比其他相更穩定。當壓力增加到31.8 GPa時,預測的正交相Pnma-ZrN2成為最穩定的相,可以通過高壓合成來制備出來。此外,在0~17.6 GPa壓力范圍內,C2/m-ZrN2相的焓值最低。在17.6~100 GPa范圍內,正交相Pnma-ZrN2的穩定性優于單斜相C2/m-ZrN2。因此,在0~100 GPa的壓力范圍內,ZrN2化合物的結構轉變順序為C2/m-ZrN2→Pnma-ZrN2。從圖2(b)中的體積-壓力關系(V-P)曲線,也可以觀察到C2/m→Pnma的相變在過渡點的體積變化不連續,體積坍縮率為10.2%,表明發生了相變。

圖2 相對焓值和體積與壓力的關系
2.2 結構特征和穩定性


注:綠色圓圈為Zr原子;金色圓圈為N原子。
為了研究ZrN2化合物的動力學穩定性,計算了0 GPa時C2/m-ZrN2,50 GPa時Pnma-ZrN2和100 GPa時Pnma-ZrN2的聲子譜,如圖4所示。由圖4可知,在整個布里淵區內沒有發現虛頻,表明ZrN2化合物的兩個結構是動力學穩定的。為了闡明ZrN2化合物在選定壓力下的力學穩定性,本文采用應力-應變關系推導出彈性常數,如表1所示。本文計算的彈性常數均很好地滿足相應空間群的晶體力學穩定性判據[19],證明了所考慮的兩個ZrN2結構在選定壓力下是力學穩定的。值得注意的是,0 GPa下Pnma-ZrN2的彈性常數也滿足晶體力學穩定性判據,則證明Pnma-ZrN2在環境條件下也具有力學穩定性。
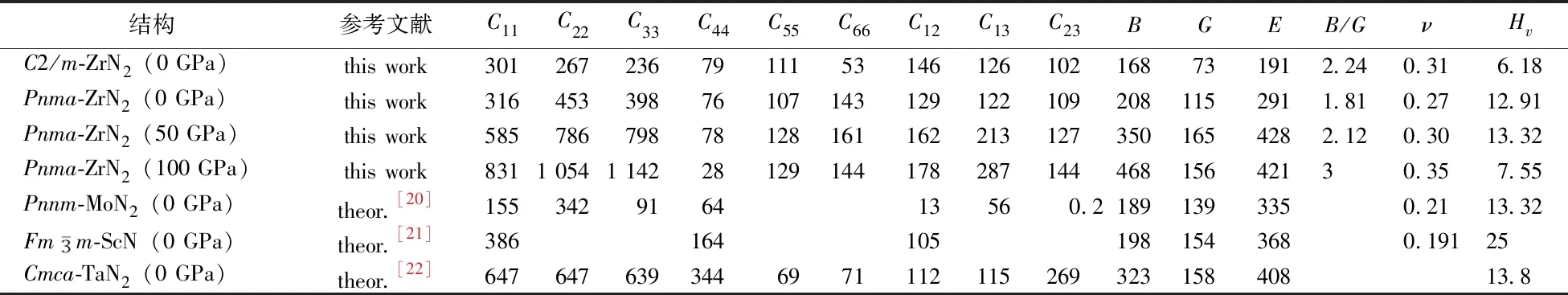
表1 ZrN2化合物在不同壓力下的力學性能

圖4 ZrN2化合物在不同壓力下的聲子譜
2.3 高壓下的力學性能和硬度
2.4 電子性質
通過對0 GPa時C2/m-ZrN2和50 GPa時Pnma-ZrN2的電子能帶結構和態密度(Density of State,DOS)進行計算(見圖5),研究ZrN2化合物的電子性質。由圖5(a)可知,C2/m-ZrN2的導帶和價帶都穿過了費米面,表明該結構具有金屬性。費米能級附近的價帶區域主要是由N-2p軌道貢獻,而導帶區域主要由Zr-4d軌道貢獻。由圖5(b)可知,50 GPa壓力下Pnma-ZrN2是一種間接帶隙的半導體化合物,其帶隙為0.615 eV。在價帶區域,主要由Zr-4d軌道和N-2p軌道的電子貢獻的;在導帶區域內,主要由Zr-4d軌道和N-2p軌道電子貢獻的。另外,ZrN2化合物的N-2p和Zr-4d軌道之間存在顯著雜化效應,可能會對ZrN2化合物的硬度產生影響。

圖5 化合物在不同壓力下的能帶結構和態密度
3 結論
1)首次發現了兩種動力學和力學穩定的新型ZrN2化合物結構:C2/m相和Pnma相,建立了ZrN2化合物在高壓下的相圖。確定了ZrN2化合物在高壓下的相變序列為C2/m→Pnma,對應的相變壓力是17.6 GPa。此外,正交相Pnma-ZrN2在31.8 GPa的壓力條件下可以通過高壓合成來制備和合成。
2)Pnma-ZrN2在0 GPa和50 GPa時硬度分別為12.91 GPa和13.32 GPa,表明ZrN2化合物是很有前途的硬質材料。
3)C2/m-ZrN2具有金屬性,Pnma-ZrN2是帶隙為0.615 eV的間接帶隙半導體。
猜你喜歡
數學雜志(2021年6期)2021-11-24 11:12:00
哲學評論(2021年2期)2021-08-22 01:53:34
中學生數理化(高中版.高二數學)(2021年5期)2021-07-21 02:14:46
數學年刊A輯(中文版)(2021年1期)2021-06-09 09:31:56
中等數學(2020年6期)2020-09-21 09:32:38
中華詩詞(2019年7期)2019-11-25 01:43:04
中等數學(2019年6期)2019-08-30 03:41:46
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·七年級數學人教版(2018年4期)2018-06-28 03:26:30
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
