
硫元素對微庫倫法測定石腦油有機氯影響規律及消除辦法研究
2023-12-06 13:11:54張韜王宗廷任亞楠盧建平張鳳梅溫聰穎盧玉坤
山東化工 2023年20期
張韜,王宗廷,任亞楠,盧建平,張鳳梅,溫聰穎,盧玉坤
(1. 中國石化勝利油田分公司勝利采油廠,山東 東營 276800;2.中國石油大學(華東) 化學化工學院化學系,山東 青島 266555)
2013年5月發生的勝利原油有機氯污染事故,再次證明了控制原油有機氯含量的極端重要性,為此中石化在國家標準GB/T 18612基礎上建立了符合油田實際的有機氯測定企業標準Q/SH 1020 2093—2016,并迅速在全國鋪開,取得了很好的應用效果[1-2],但也出現了很多問題。
在原油開采過程中后期,油田三次采油需要使用的各種含有機氯的化學助劑如清蠟劑、降粘劑、瀝青分散劑、破乳劑等,這些有機氯化物來源與組成十分復雜,逐一定性和定量分析非常困難,只能采用氧燃燒法使這類有機氯在火焰中分解生成氯化氫,通過電解液吸收后測定氯離子含量,間接測出有機氯總量。
在標準執行過程中,由于原油來源與組成十分復雜,加之測定儀器眾出多家,操作規程不統一等問題是客觀現實的存在,測定中數值不準確,甚至差別極大(相對偏差50%以上)[3-6],重復性也很差,特別是有機氯含量低于5 μg/g時精密度非常差。開展影響測定因素研究,就顯得必要而緊迫。由于整個測定過程從原油蒸餾、洗滌、進樣、霧化、燃燒裂解、吸收、微庫倫滴定等程序,操作流程非常長,需要逐個進行研究和優化。
原油有機氯測定標準中特別提到硫元素的影響,總硫氯比大于10 000時會給測定帶來嚴重影響。原油中硫的存在形式主要是硫化氫、硫醇、硫醚以及含硫芳香族化合物等,盡管硫的存在形式多樣,但氧燃燒后主要產物為二氧化硫,不必區分不同形式的硫的分別影響規律,只需考慮硫含量大小對有機氯測定的影響即可。故本論文主要研究了不同含量硫元素對不同含量有機氯測定的影響及影響規律,并分析了影響發生機理,并提出了相應的解決辦法。改進后的辦法大大抑制了硫的影響程度,有機氯測定的準確度、精密度都得到明顯的提高。
1 實驗部分
1.1 主要儀器與試劑
1.1.1 主要儀器
WC-2000微庫侖儀,江蘇江分電分析儀器有限公司;
石腦油蒸餾儀,山東德豐石油裝備有限公司;
微量注射器,10 μL,上海安亭微量進樣器廠;
電解池陽極為工作電極:涂膜銀電極,陰極為對電極:鉑絲電極;
原電池指示電極為涂膜銀電極,參比電極為雙鹽橋飽和硫酸亞汞電極。
1.1.2 主要試劑
銀電極動態保護劑(T試劑,主要成分為苯駢三氮唑及其衍生物的混合物),自制,上海奧克林生化有限公司;
質量分數2%T試劑-冰醋酸溶液;
冰醋酸,分析純,天津分析試劑有限公司;
體積分數70%冰醋酸水溶液;
氯含量測定用標準物質,10 μg/mL,石油化工科學研究院;
氯含量測定用標準物質,0.50 μg/mL,石油化工科學研究院;
硫含量測定用標準物質,2 000 μg/mL,石油化工科學研究院;
二甲基二硫,分析純,J&K Chemical。
1.2 實驗方法
1.2.1 有機氯測定方法
1.2.1.1 標準有機氯測定方法
WK-2000 型微庫侖綜合分析儀的參數設置 :積分電阻 2 kΩ、放大倍數2 000、偏壓 105~150 kV。
先收集原油204 ℃(稠油320 ℃)前石腦油餾分,經堿洗除硫化氫、三次水洗除鹽后,離心脫水后,抽取6.4 μL(或適量)石腦油注入氧燃燒器充分燃燒后,轉化成鹽酸,被15 mL 70%乙酸溶液吸收后,電生銀離子Ag+,生成AgCl沉淀,消耗掉吸收的氯離子Cl-,通過消耗的電量換算出石腦油中有機氯含量。
氯化物在滴定池中的反應如下:
Cl-+Ag+→ AgCl ↓
上述反應中消耗的銀離子發生的庫侖反應如下:
Ag→Ag++e-
1.2.1.2 標準改進型有機氯測定方法
先收集原油204 ℃(稠油320 ℃)前石腦油餾分,經堿洗除硫化氫、三次水洗除鹽,離心脫水后,抽取6.4 μL(或適量)石腦油注入氧燃燒器充分燃燒后,轉化成鹽酸,被15 mL70%乙酸溶液(內添加3滴電極動態修飾劑T試劑)吸收鹽酸后,電生銀離子Ag+,生成AgCl沉淀,消耗掉吸收的氯離子Cl-,通過消耗的電量換算出石腦油中有機氯含量。
1.2.2 不同含量硫元素對有機氯測定干擾規律測定方法
1.2.2.1 以二甲基二硫(DMDS) 配制含硫樣品的方法(Ⅰ)
1)取1.0 mL二甲基二硫(DMDS) 加入10 mL容量瓶中,取6.4 μL,測其有機氯。
(分子量M=94.2,d=1.062 5,cS=7.23×105μg/mL),注意觀察是否出峰、峰形。
2)在上述1瓶中,加入1.0 mL氯標準溶液(cCl=10 μg/mL),混勻,總體積2.0 mL,測其有機氯。
此時,cCl=5.0 μg/mL,cS=3.62×105μg/mL,cS/cCl=7.23萬倍。
3)在上述2瓶中,再加入1.0 mL氯標,混勻,總體積3.0 mL,測其有機氯。
此時,cCl=6.7 μg /mL,cS=2.41×105μg/mL,cS/cCl=3.61萬倍。
4)在上述3瓶中,再加入1.0 mL氯標,混勻,總體積4.0 mL,測其有機氯。
此時,cCl=7.5 μg /mL,cS=1.80×105μg/mL,cS/cCl=2.40萬倍。
5)在上述4瓶中,再加入1.0 mL氯標,混勻,總體積5.0 mL,測其有機氯。
此時,cCl=8.0 μg /mL,cS=1.45×105μg/mL,cS/cCl=1.80萬倍。
6)在上述5瓶中,再加入5.0 mL氯標,混勻,總體積10.0 mL,測其有機氯。
此時,cCl=9.0 μg /mL,cS=7.23×104μg/mL,cS/cCl=8 033倍。
1.2.2.2 以二甲基二硫(DMDS) 配制含硫樣品的方法(Ⅱ)
1)取1.0 mL二甲基二硫(DMDS)加入10.0 mL容量瓶中,取6.4 μL,測其有機氯。
(分子量M=94.2,d=1.062 5,cS=7.23×105μg/mL),注意觀察是否出峰,峰形,沉淀顏色及顆粒大小。
2)在上述1瓶中,加入1.0 mL氯標(質量濃度30 μg/mL ),混勻,總體積2.0 mL,測其有機氯。此時,cCl=15.0 μg/mL,cS=3.62×105μg/mL,cS/cCl=2.413萬倍。
3)在上述2瓶中,再加入1.0 mL氯標,混勻,總體積3.0 mL,測其有機氯。
此時,cCl=20.0 μg/mL,cS=2.41×105μg/mL,cS/cCl=1.21萬倍。
4)在上述3瓶中,再加入1.0 mL氯標,混勻,總體積4.0 mL,測其有機氯。
此時,cCl=22.5 μg/mL,cS=1.80×105μg/mL,cS/cCl=8 000倍。
5)在上述4瓶中,再加入1.0 mL氯標,混勻,總體積5.0 mL,測其有機氯。
此時,cCl=24.0 μg/mL,cS=1.45×105μg/mL,cS/cCl=6 042倍。
6)在上述5瓶中,再加入5.0 mL氯標,混勻,總體積10.0 mL,測其有機氯。
此時,cCl=27.0 μg/mL,cS=7.23×104μg/mL,cS/cCl=2 678倍。
1.2.2.3 以硫標準溶液配制含硫樣品的方法(Ⅲ)
1)取1.0 mL硫標準物質(cs=2 000 μg/mL),加入0.40 mL氯標準物質(cCl=0.50 μg/mL)充分混合均勻后,cS/cCl=1萬倍,測量有機氯 。
2)在上述1瓶中,再加入0.10 mL氯標準物質充分混合均勻后,cS/cCl=0.8萬倍;
3)在上述2瓶中,再加入0.17 mL氯標準物質充分混合均勻后,cS/cCl=0.6萬倍;
4)在上述3瓶中,再加入0.33 mL氯標準物質充分混合均勻后,cS/cCl=0.4萬倍;
5)在上述4瓶中,再加入1.0 mL氯標準物質充分混合均勻后,cS/cCl=0.2萬倍。
1.2.2.4 以硫標準溶液配制含硫樣品的方法(Ⅳ)
1)取1 mL硫標準物質,加入0.2 mL氯標準物質充分混合均勻后,cS/cCl=2萬倍;
2)在上述1瓶中,再加入22.2 μL氯標準(同Ⅲ)物質充分混合均勻后,cS/cCl=1.8萬倍;
3)在上述2瓶中,再加入27.8 μL氯標準物質充分混合均勻后,cS/cCl=1.6萬倍;
4)在上述3瓶中,再加入35.7 μL氯標準物質充分混合均勻后,cS/cCl=1.4萬倍;
5)在上述4瓶中,再加入47.6 μL氯標準物質充分混合均勻后,cS/cCl=1.2萬倍。
2 實驗結果與討論
2.1 以二甲基二硫(DMDS) 配制含硫樣品有機氯結果(Ⅰ)
用氧燃燒法測定石腦油有機氯含量時,硫元素的存在都會產生一定的影響,參照標準方法重點考察了不同硫氯含量比對有機氯測定的影響規律。首先我們選擇了含硫量比較大的二甲基二硫(DMDS)作為研究對象,測得了一系列不同硫氯比下有機氯的測定結果,見表1,測定結果表明,以70%乙酸水溶液為底液時,純的DMDS也顯示出較高的有機氯測定值,為2.8 μg/mL,筆者認為有兩種可能,一是試劑中存在一定濃度的有機氯,二是燃燒氧化生成的二氧化硫被電解液吸收生成亞硫酸,在陽極上電解生成SO42-,消耗一定的電量,對測定產生較大的干擾。但在測定含氯樣品時,發現測定值比理論氯含量高,也比計算氯含量高(將DMDS中的氯也加進去),且增加值幅度變化很大(從+0.2~2.5 μg/mL不等)。故筆者認為既有試劑中有機氯的影響,也有二氧化硫電解造成的干擾問題,cS/cCl比改變沒有發現明顯的影響規律。
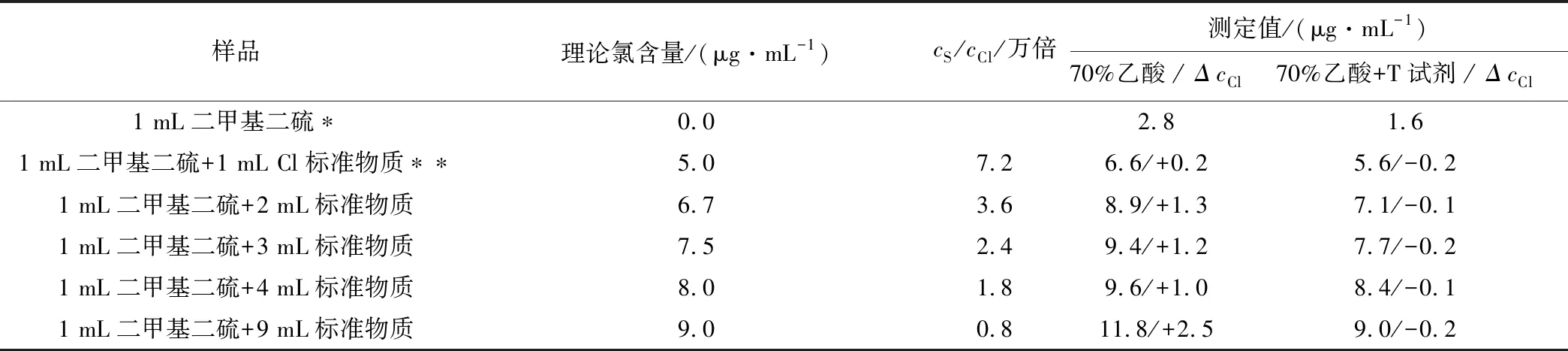
表1 元素硫影響下有機氯測定結果(Ⅰ)
當在70%乙酸電解液中加入電極動態修飾T試劑時,發現空白DMDS有機氯測定值減小到1.6 μg/mL,且測定值與計算氯含量非常接近,稍微偏低,減小值變化幅度很小(從-0.1~到-0.2 μg/mL)。筆者驚喜地發現,T試劑基本上抑制了硫的干擾問題。
2.2 以二甲基二硫(DMDS) 配制含硫樣品有機氯結果(Ⅱ)
為了進一步研究硫元素對有機氯測定的影響規律,又選取了濃度為30 μg/mL的有機氯標準物質,以減小硫氯比,測定結果見表2。從表2可以看出, 在70%乙酸介質中,測定值普遍偏低,變化幅度也比較大(從-0.9到-2.2 μg/mL),但變化幅度相對值變小,說明有機氯含量高時,測定誤差也變小,即二氧化硫造成的影響減小。跟表1比較后發現,硫的影響由正變負,原因可能是有機氯燃燒不完全所致,也有可能是二氧化硫的存在,使指示電極發生生成銀離子的化學反應,導致指示電極工作失常,無法正確指示平衡電位。實驗中也發現,測含硫試樣后,偏壓值很難調,基線容易飄移,結果重現性變差,每一次測定,結果都發生變化,也就是硫元素對測定的影響非常嚴重。cS/cCl比改變沒有發現明顯的影響規律,也就硫氯比不同,即便硫氯比3 000,產生的影響是相似的,主要原因可能還是指示電極工作失常所致。
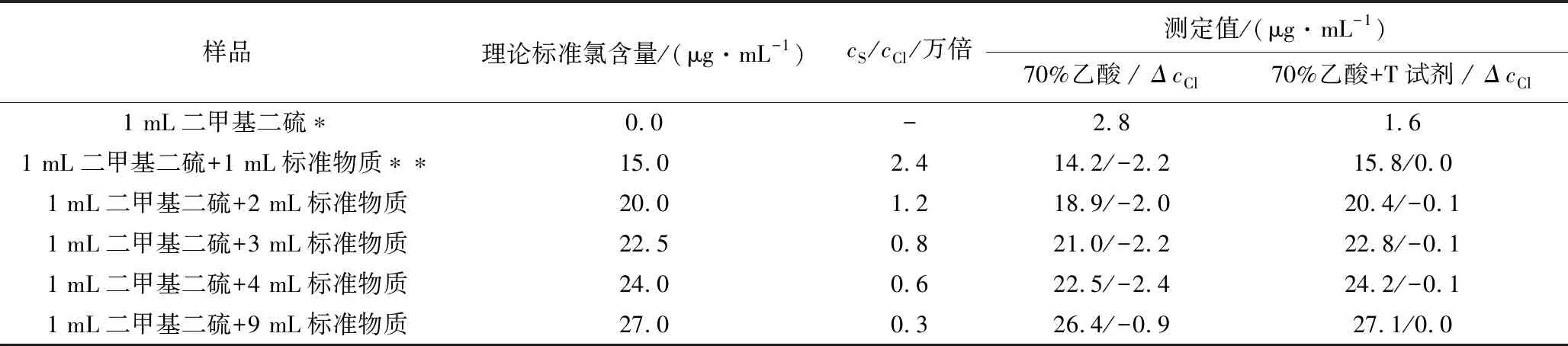
表2 元素硫影響下有機氯測定結果(Ⅱ)
當在70%乙酸電解液中加入電極動態修飾T試劑時,測定值與計算氯含量非常接近,稍微偏低,減小值變化幅度很小(從0.0到-0.1 μg/mL)。筆者同樣發現,T試劑基本上抑制了硫的干擾問題,作用機理是既抑制了二氧化硫在工作電極上的電解,也保護了指示電極的零電流工作狀態,取得比較理想的測定結果。即便硫氯比為2.4萬,由于絕對氯含量較高(15~27 μg/mL),測定誤差也很小。
2.3 以硫標準溶液配制含硫樣品的有機氯測定結果(Ⅲ)
由于以上兩種實驗硫的絕對濃度很高,導致電極工作狀態不正常,加之其自身的有機氯含量無法準確測定,故改用元素硫標準物質(cS=2 000 μg/mL)作為硫元素來源,降低硫元素的絕對濃度,降低其不利影響,并且與實際石腦油樣品含硫濃度相似,配置了一系列不同硫氯比的含氯試樣,測定有機氯結果見表3。

表3 元素硫影響下有機氯測定結果(Ⅲ)
從表3可以看出,在70%乙酸介質中,硫標準物質也表現出含有有機氯,有機氯測定值遠高于理論氯標準物質濃度,且數值忽大忽小,完全看不到有機氯的濃度變化規律。如果把空白硫標準物質的表觀有機氯算進去,硫影響也是忽負忽正,難以找到規律。
但在70%乙酸加T試劑的試液中,空白硫標液表現出有機氯測定值,但僅為標準方法的18%,其余含氯試樣其測定值與理論氯標基本一致,表現出較好的規律性。即便把空白硫標中的表觀有機氯算進去,硫元素影響普遍表現為負偏差,變化幅度基本一致。
有一種可能就是空白硫標樣不含有機氯,其測定值是一種假性結果,即由于二氧化硫電解所消耗電量所致。
當在電解液含氯離子時,由于銀離子迅速跟氯離子反應,降低了銀離子的析出電位,在與二氧化硫電解競爭中勝出,只有當氯被電解生成的銀離子反應完后,才能輪到二氧化硫,而此時二氧化硫也基本被快速流動的氣流帶走了,其影響可以忽略不計。
因此,在70%乙酸介質中,如果假設空白硫標樣不含有機氯,但有機氯測定結果卻比理論有機氯大很多,數據完全失真。筆者認為原因不是出在工作電極上,而是出在指示電極上,由于二氧化硫的存在,使指示電極本身銀單質發生了電化學反應,在氧氣存在的條件下,電解生成銀離子,也就是無法保持零電流的測定條件,測定結果也會失真。
2.4 以硫標準溶液配制含硫樣品的有機氯測定結果(Ⅳ)
為進一步研究高硫氯比下,對超低有機氯含量的影響規律,參照以硫標準溶液配制含硫樣品的方法(Ⅳ)配置一系列不同硫氯比的超低有機氯含量樣品,測試其有機氯含量,結果見表4。
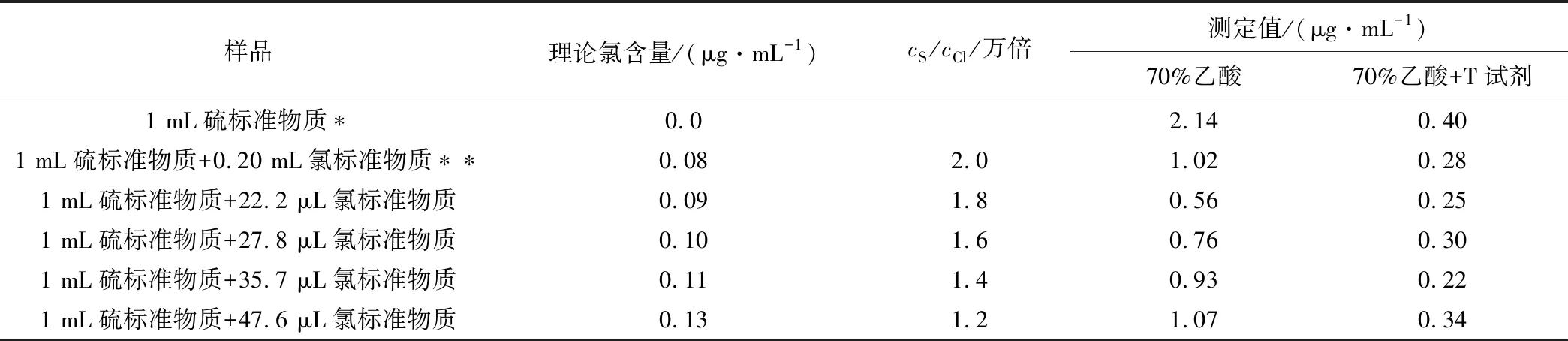
表4 高硫氯比、超低有機氯含量測定結果(Ⅳ)
根據測定標準,本方法的適宜測定范圍為0.5~25 μg/mL,當氯含量在0.1 μg/mL時,結果將出現很大誤差,一般認為測定誤差在100%也是允許的。本表4所涉及的氯含量在0.08~0.13之間,且硫氯比在1.2萬以上,誤差一定會大幅度增加。
盡管空白硫標準物質表現出較高的有機氯測定值,根據前面的分析,仍然假設有機氯含量為零。在70%乙酸介質中,硫氯比1.2萬以上,有機氯的絕對含量超低,誤差高達600%~1 300%,且大多在600%~800%之間,影響程度變化不大,說明二氧化硫在電解競爭中處于優勢地位。反觀在70%乙酸介質+T試劑中,誤差在200%~300%之間,與標準電解質相比,誤差大幅度降低,但硫元素的影響還是比較嚴重的。也進一步證實了硫氯比高于1萬時,其影響不能忽略。
3 結論
硫元素的存在確實會對原油(石腦油)有機氯測定產生影響,不僅會使測定結果產生很大的測定誤差,而且精密度變差,基線不穩且發生偏移。電解液中加入T試劑可以在工作電極與指示電極銀電極保護膜的基礎上進行動態修飾,既可對防止銀單質的氧化酸解腐蝕帶來的不利影響(非陽極電解產生的銀離子,以及指示電極偏離平衡狀態、偏壓改變),又可以增大二氧化硫的電極氧化過電位,抑制其電解產生的電量測定誤差。通過實驗得出如下結論:
1)按標準測定方法,硫元素的存在對有機氯(含量在標準范圍0.50~25μg/mL以內)的測定產生±5%~±25%的測定誤差,且誤差出現的規律性很差。即便硫氯比為2 000時,也產生較大的影響。原因主要表現在兩個方面:一是燃燒氧化生成的二氧化硫被電解液吸收生成亞硫酸,在陽極(工作電極)上電解生成SO42-,消耗一定的電量,對測定產生較大的干擾;二是在指示電極上,由于二氧化硫的存在,使指示電極本身銀單質發生了電化學反應,電解生成的銀離子與亞硫酸根生成亞硫酸銀配合物,氧氣還原為水,指示電極表面電極電位發生偏移,也就是無法保持零電流的測定條件,測定結果也會產生較大誤差。并且發現,影響程度并沒有隨硫氯比增大而增大,忽大忽小,沒有明顯的影響規律;但隨著有機氯濃度的增加相對誤差會變小,也就是影響會變小,二氧化硫在陽極上的電解處于競爭中的劣勢。
2) 按改進后的標準方法,在電解液中加T試劑對工作電極和指示電極動態修飾后,有機氯測定誤差+0%-+3%之間,硫氯比盡管很大(0.3萬~7.2萬),其影響仍然很小,且誤差大小基本恒定,測定結果基本接近正常值。
T試劑的作用機制是在工作電極與指示電極銀電極保護膜的基礎上,對裸露的銀活性位點進行吸附性保護覆蓋,既可對防止銀單質的氧化酸解腐蝕帶來的不利影響(非陽極電解產生的銀離子,以及指示電極偏離平衡狀態、偏壓改變),又可以增大二氧化硫的電極氧化過電位,抑制其電解產生的電量測定誤差。
3)按標準測定方法,硫元素的存在對有機氯(含量小于0.50 μg/mL以內)的測定產生+600%~+1 300%的測定誤差,且誤差出現的規律性很差。即便硫氯比為2 000時,也產生較大的影響。原因可能是在70%乙酸介質中,硫氯比1.2萬以上,有機氯的絕對含量超低,二氧化硫在電解競爭中處于優勢地位。
4)按改進后的標準方法,對低有機氯含量的測定產生100%測定結果是允許的,但由于硫元素的影響,誤差在+200%~+300%之間,與標準方法相比,誤差雖大幅度降低,但硫元素的影響還是比較嚴重的。也進一步證實了硫氯比高于1萬時,其影響不能忽略。原因可能是有機氯含量低時,銀離子的析出電位增高,達到了二氧化硫的氧化電位,電解產生的電量誤差就不可避免了。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·八年級物理人教版(2022年3期)2022-03-16 05:55:08
當代陜西(2021年2期)2021-03-29 07:41:24
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
媽媽寶寶(2017年3期)2017-02-21 01:22:28
中國塑料(2016年3期)2016-06-15 20:30:00
通信電源技術(2016年3期)2016-03-26 07:13:38
專用汽車(2016年4期)2016-03-01 04:13:43
