敲低膜聯蛋白A2對食管鱗癌細胞惡性表型的抑制作用
2023-12-12 07:32:14王意濃劉國政謝觀超王明榮郝佳潔
癌變·畸變·突變 2023年6期
王意濃,劉國政,謝觀超,馮 丹,李 賽,蔡 巖,張 鈺,王明榮,郝佳潔
(國家癌癥中心/國家腫瘤臨床醫學研究中心/中國醫學科學院北京協和醫學院腫瘤醫院,分子腫瘤學全國重點實驗室,北京 100021)
食管癌位居全球癌癥死因第6位[1];中國作為全球食管癌發病風險最高的地區之一,每年食管癌新發病例數約占全球的53.7%。其中,食管鱗癌病例數占食管癌病例總數的90.4%[2]。腫瘤復發和轉移是食管鱗癌相關死亡的主要原因,我國食管鱗癌早期診斷率低,總體預后比較差[3]。因此,尋找食管鱗癌新的診斷標志物和潛在治療靶點,對食管鱗癌患者的治療以及預后十分重要。
膜聯蛋白家族(annexins,ANXs)在動物細胞和植物細胞中廣泛表達,目前已鑒定出超過160 個成員。ANXA2作為該家族蛋白中最主要的成員之一,已被證明在多種惡性腫瘤中高表達,并與腫瘤的分期及預后相關[4]。本課題組之前的研究發現,ANXA2 在食管鱗癌細胞系中高表達,使用siRNA瞬時敲降ANXA2后顯著減弱食管鱗癌細胞的侵襲遷移和肺轉移能力;機制研究表明,ANXA2 可以通過上調MYC 蛋白表達進而促進食管鱗癌細胞的侵襲遷移[5]。
CRISPR/Cas9 系統包含規律成簇的間隔短回文重復序列(clustered regularly interspaced short palindromic repeats,CRISPR)和具有切割DNA雙鏈作用的CRISPR相關蛋白9(CRISPR-associated protein 9,Cas9)組成,是由RNA 指導Cas9 核酸酶編輯靶向基因的技術[6]。CRISPR/Cas9 系統由向導RNA(guide RNA,gRNA)和Cas9 蛋白形成復合體,gRNA 與目標基因的靶序列通過堿基互補配對原則進行配對,Cas9蛋白在目標DNA序列的特異性識別位點進行定點切割,從而導致雙鏈斷裂[7]。CRISPR 編輯技術已在臨床前和臨床研究中使用,用于有害突變基因的敲除和錯誤編碼序列的修復[8-9],使用該技術還可對腫瘤細胞中高表達的基因進行敲除[9],為腫瘤治療的基礎研究和臨床轉化奠定基礎。與傳統的RNA干擾技術相比,該系統可產生穩定的基因敲除,并且脫靶效應也明顯減少[10]。
本研究使用CRISPR/Cas9 基因編輯技術,借助慢病毒載體敲低食管鱗癌KYSE30 和KYSE150 細胞中的ANXA2基因,并初步探索了敲低ANXA2后食管鱗癌細胞表型的變化,為深入研究ANXA2 的生物學功能及其高表達促進食管鱗癌細胞侵襲遷移的作用機制提供了技術基礎。
1 材料與方法
1.1 細胞培養
食管鱗癌細胞KYSE30 和KYSE150 均由日本東京大學Shimada 教授惠贈,分別使用含有10%胎牛血清(Newzerum)的RPMI-1640 培養基(北京細工生物公司),置于CO2體積分數為5%的培養箱(美國Thermo Fisher Scientific 公司)中培養。人胚腎細胞HEK293FT(美國Invitrogen 公司)使用含10%胎牛血清的DMEM 高糖培養基(北京細工生物公司),添加1% L-型谷氨酰胺、1%非必需氨基酸以及1% G418(美國Gibco 公司),置于培養箱中培養。
1.2 質粒構建
1.2.1 gRNA 靶點及寡核苷酸鏈序列在Addgene網站中查詢及下載文獻[11-12]中報道的ANXA2基因及對照CRISPR gRNA(https://www.addgene.org/pooled-library/zhang-human-gecko-v2/),在正義鏈模板的5'端添加CACCG,反義鏈模板的5'端添加AAAC,與BsmBI 酶切后形成的黏性末端互補。選取了ANXA2 gRNA 共6對寡核苷酸,序列見表1。

表1 對照及ANXA2 gRNA寡核苷酸單鏈名稱及序列
1.2.2 gRNA 表達質粒的構建將合成的gRNA 正義鏈和反義鏈粉末(北京天一輝遠生物科技有限公司)稀釋至100 μmol/L。退火體系:10×T4 DNA Ligase Buffer(NEB) 1 μL,正義鏈1 μL,反義鏈1 μL,T4 PNK(NEB) 0.5 μL, ddH2O 6.5 μL。 退 火 條 件:37 ℃、30 s;95 ℃、5 min;5 ℃/min 梯度降溫至25 ℃,-20 ℃保存。然后用BsmBI 酶切載體:10×NEB Buffer 5 μL、pLenti-U6-gRNA-Cas9-P2A-EGFPPuro&Zeocin 載體(簡稱pLenti-U6-gRNA-Cas9 載體;上海碧云天生物技術有限公司) 3 μg、BsmBI-v2(NEB)1 μL、加ddH2O 至50 μL。酶切條件:55 ℃、14 min;80 ℃、20 min。接著使用瓊脂糖凝膠DNA回收試劑盒(天根生化科技有限公司)回收酶切片段。
下一步進行酶切載體與雙鏈gRNA 連接:酶切載體50 ng,2×Quick Ligase Buffer(NEB) 5 μL,雙鏈gRNA 1 μL,加ddH2O 至10 μL,最后加入1 μL Quick Ligase,25 ℃、15 min。最后進行連接產物轉化及鑒定:將連接產物轉化至Stbl3感受態細胞(北京全式金生物技術股份有限公司),均勻涂布至含100 μg/mL 氨芐青霉素的固體LB 培養基,37 ℃過夜培養,每個靶點挑取3 個單菌落,加入含100 μg/mL 氨芐青霉素的液體LB 培養基,200 r/min,37 ℃震蕩培養過夜。菌液經PCR、瓊脂糖凝膠電泳及Sanger 測序驗證(測序引物5'-CAAGGCTGTTAGAGAGATAA-3',由北京天一輝遠生物科技有限公司完成)。測序完成后,使用質粒提取試劑盒(北京康為世紀生物科技有限公司)提取質粒。
1.3 慢病毒制備
將HEK293FT細胞接種至6 cm平皿中,待細胞密度長至50%~70%。將CRISPR質粒6 μg、慢病毒包裝輔 助 載 體psPAX2 和pMD2G 各3 μg 混 合,按 照JetPRIME 轉 染 試 劑 說 明 書(PolyPlus 公 司) 轉 染HEK293FT 細胞,48 h 后收集細胞培養上清,0.22 μm 過濾器過濾除菌,使用病毒滴度檢測卡(北京博奧龍免疫技術有限公司)檢測病毒滴度,保存于-80 ℃冰箱備用。
1.4 慢病毒感染細胞
分別將KYSE30 和KYSE150 細胞接種至96 孔板,待細胞密度長至30%~50%,根據病毒滴度加入含70%病毒培養上清的培養基。慢病毒、25×感染增強試劑P(上海吉凱基因醫學科技股份有限公司)進行感染,感染20 h 后更換為RPMI-1640 培養基繼續培養。培養48 h后,使用含2 μg/mL嘌呤霉素(上海碧云天生物技術有限公司)的RPMI-1640培養基篩選3~4 d。
1.5 Western blot實驗
分別收集KYSE30 和KYSE150 細胞加入胰蛋白酶(美國Thermo Fisher Scientific 公司)進行消化,1 000 r/min離心5 min,棄上清獲得細胞沉淀,使用RIPA裂解液(上海碧云天生物技術有限公司)提取細胞蛋白,使用BCA 蛋白定量試劑盒(美國Thermo Fisher Scientific公司)進行定量,取15 μg蛋白樣品,加入5×上樣緩沖液(上海碧云天公司),98 ℃金屬浴變性10 min。聚丙烯酰胺凝膠電泳90 min,使用濕轉電轉儀將蛋白從分離膠轉至PVDF膜上,5%脫脂奶粉室溫封閉1 h,加入一抗后4 ℃孵育過夜,一抗包含:ANXA2 抗 體(Abcam 公 司,ab189473)、MYC 抗 體(Abcam 公 司,ab32072)、GAPDH 抗 體(Cell Signaling Technology 公司,2118);使用洗滌緩沖液TBST 洗膜4次,每次6 min,加入山羊抗兔二抗(北京中杉金橋,ZB-2301),室溫孵育1 h,使用洗滌緩沖液TBST洗膜4 次,每次6 min,使用ECL 化學發光液(美國Thermo Fisher Scientific公司)進行顯影、曝光。
1.6 總RNA提取和實時定量PCR檢測
使用RNA 提取試劑盒(北京康為世紀生物科技有限公司)提取KYSE30細胞總RNA,將得到的RNA作為模板,使用HiFi Script cDNA 合成試劑盒(北京康為世紀生物科技有限公司)逆轉錄合成cDNA。使用TB Green?Premix ExTaqTM試劑盒(日本TaKaRa 公司)進行逆轉錄實時熒光定量PCR(reverse transcription quantitative real-time PCR,RT-qPCR)實驗,mRNA相對表達量以2-ΔΔCT表示,具體操作步驟參照試劑說明書進行。ANXA2 及內參GAPDH 引物分別為:ANXA2,上游5'-GAGCGGGATGCTTTGAACATT-3',下游5'-TAGGCGAAGGCAATATCCTGT-3';GAPDH,上游5'-GGAGCGAGATCCCTCCAAAAT-3',下游5'-GGCTGTTGTCATACTTCTCATGG-3'。
1.7 細胞遷移與侵襲實驗
1.7.1 遷移實驗從24 孔板取出Transwell 小室(美國Corning 公司),向孔內加入700 μL 含20%血清的RPMI-1640培養基,向小室內加入200 μL不含血清的培養基,室溫放置10 min后棄去液體。細胞消化離心后使用PBS 清洗細胞,重懸并計數。向小室內分別加入KYSE30和KYSE150細胞5×104個(體積200 μL),置于培養箱內培養12~24 h。取出上室,用無菌棉球擦拭上室,使用PBS清洗兩次,晾干后加入固定液(甲醇和丙酮體積比1∶1)固定20 min,棄去固定液,加入甲醇稀釋的0.5%結晶紫染色30 min,流水清洗。晾干,滴加樹脂后用蓋玻片封片,鏡下拍照,計數。
1.7.2 侵襲實驗將基質膠(美國Corning公司)用無血清無抗生素RPMI-1640 培養基按照1∶33 的比例稀釋,取50 μL 加至Transwell 小室中,室溫靜置1 h,其余操作同細胞遷移實驗。
1.8 細胞集落形成實驗
收集KYSE30 細胞消化后計數,將1 000 個細胞均勻接種于6 孔板中,使用RPMI-1640 培養基于37 ℃、CO2體積分數為5%的培養箱中培養1周后,棄掉培養基,使用甲醇固定30 min,結晶紫染色10 min,拍照并計數集落數量。
1.9 統計學處理
以上所有實驗均獨立重復3 次,采用GraphPad Prism 8.0軟件進行數據統計分析。兩組間數據比較采用獨立樣本t檢驗,多組間數據比較采用單因素方差分析,P<0.05為差異具有統計學意義。
2 結 果
2.1 ANXA2基因敲低gRNA表達質粒的構建
pLenti-U6-gRNA-Cas9-P2A-EGFP-Puro&Zeocin質粒具有2 個BsmBI 內切酶的識別位點,位點之間為相距1 885 bp的填充序列(圖1A)。將ANXA2的6對經退火處理的gRNA 寡核苷酸雙鏈與酶切后的載體連接,轉化Stbl3感受態細菌,篩選出陽性克隆。對照及各靶點單克隆菌液Sanger 測序結果顯示,對照及6 個靶點序列均正確插入pLenti-U6-gRNA-Cas9 載體,插入序列的位置、方向及序列與理論一致(圖1B),表明對照及ANXA2基因敲低gRNA表達質粒構建成功。
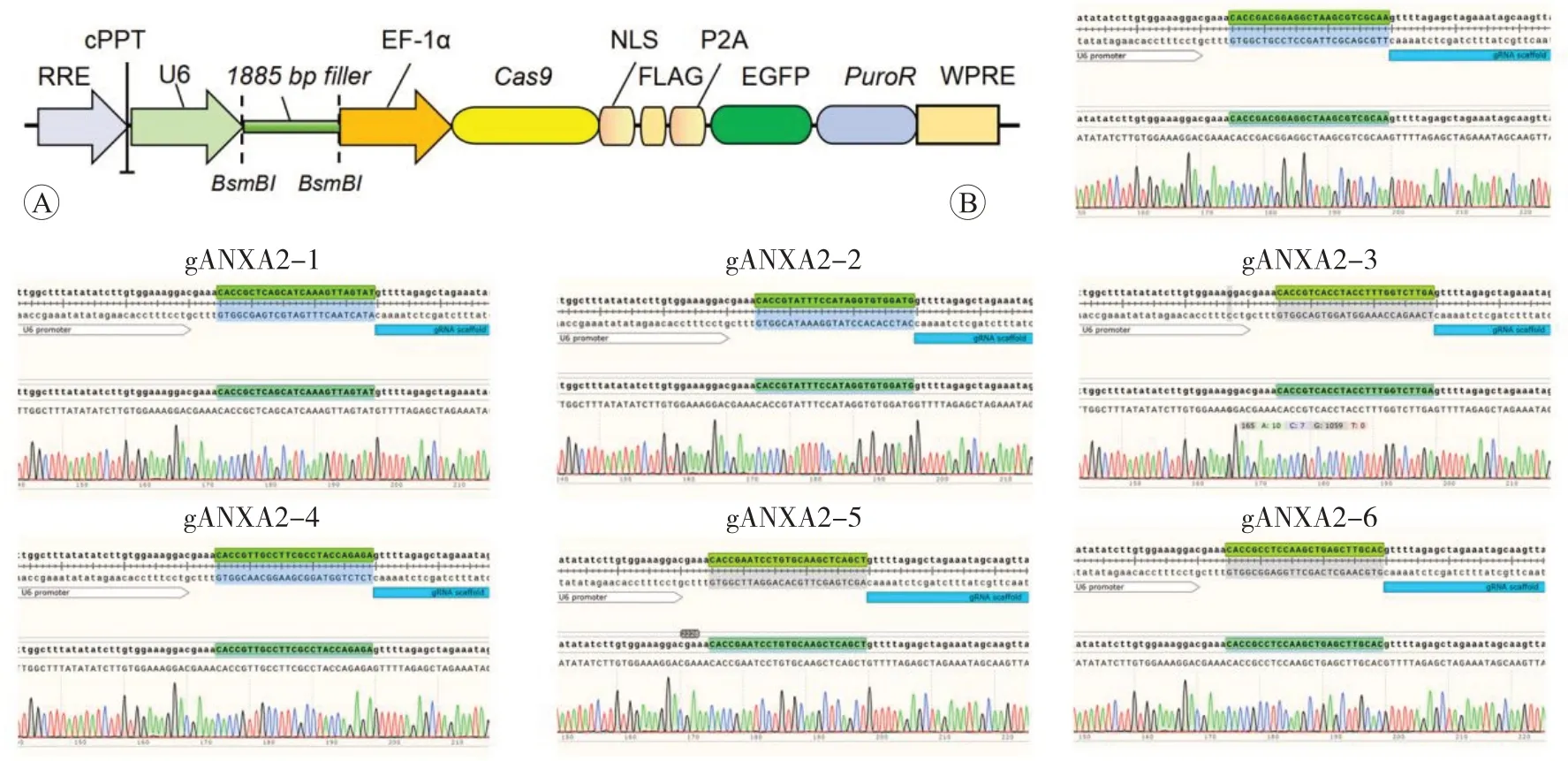
圖1 對照及ANXA2基因敲低gRNA表達質粒的構建
2.2 穩定敲低細胞中ANXA2的表達顯著降低
鑒于ANXA2 在食管鱗癌KYSE30 中表達水平較高,我們使用該細胞進行病毒感染及嘌呤霉素篩選,以獲得敲低ANXA2 的KYSE30 細胞。利用Western blot 檢測了細胞中ANXA2 蛋白表達情況,結果見圖2,顯示具有gRNA 靶點4~6 的細胞中ANXA2 的蛋白水平較對照顯著降低,而在具有gRNA 靶點1~3 的細胞中未見明顯改變。Western blot 結果還顯示,具有ANXA2 敲低gRNA 靶 點5 和6 的KYSE30 細 胞中MYC蛋白的表達顯著降低,與此前使用siRNA 敲降[5]的結果一致,而靶點1~4 gRNA中MYC蛋白水平未見明顯變化。具有gRNA 靶點5 和6 的穩定敲低細胞中ANXA2的mRNA水平顯著降低。以上結果表明,穩定敲低ANXA2基因的KYSE30細胞系構建成功,且gRNA靶點5 和6 的細胞中ANXA2 和MYC 均顯著下調。因此,后續實驗均使用這兩個靶點進行。
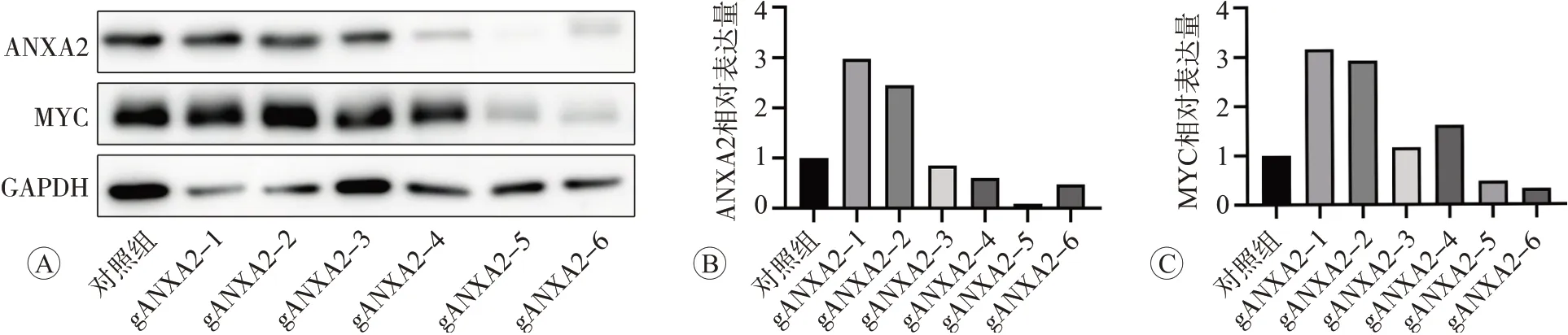
圖2 ANXA2敲低gRNA靶點的篩選
2.3 敲低ANXA2 顯著降低食管鱗癌細胞的遷移侵襲和集落形成能力
我們進一步增加構建了食管鱗癌細胞KYSE150的ANXA2 穩定敲低細胞(圖3 和圖4),并檢測了敲低ANXA2 對 食 管 鱗 癌KYSE30 和KYSE150 細 胞 的ANXA2 蛋白和mRNA 表達水平以及對細胞表型的影響。結果顯示,與對照組相比,敲低ANXA2 后,食管鱗癌KYSE30 和KYSE150 細胞的ANXA2 蛋白和mRNA 表達水平均顯著降低(P<0.05 或P<0.01),而且兩種細胞的遷移、侵襲能力均顯著下降(均為P<0.01,圖5),KYSE30 細胞的集落形成能力顯著減弱(P<0.01,圖6),提示食管癌細胞的惡性表型受到了明顯抑制。

圖3 Western blot檢測ANXA2敲低細胞中ANXA2蛋白表達情況
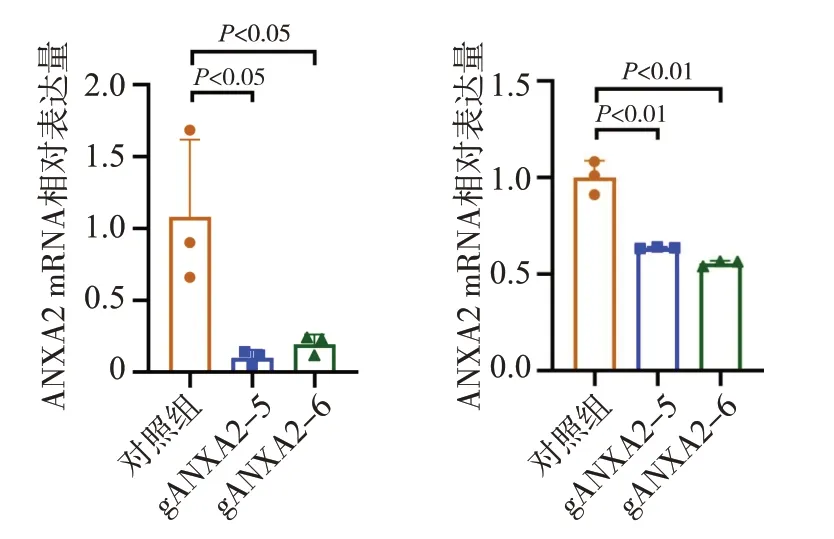
圖4 qPCR檢測ANXA2敲低細胞中ANXA2 mRNA表達情況
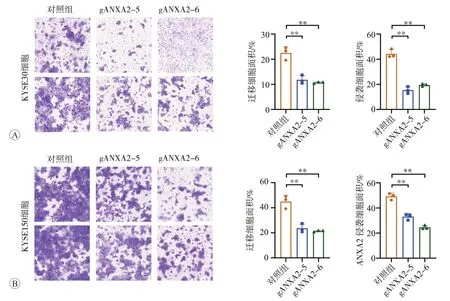
圖5 Transwell實驗檢測ANXA2敲低對細胞遷移和侵襲能力的影響
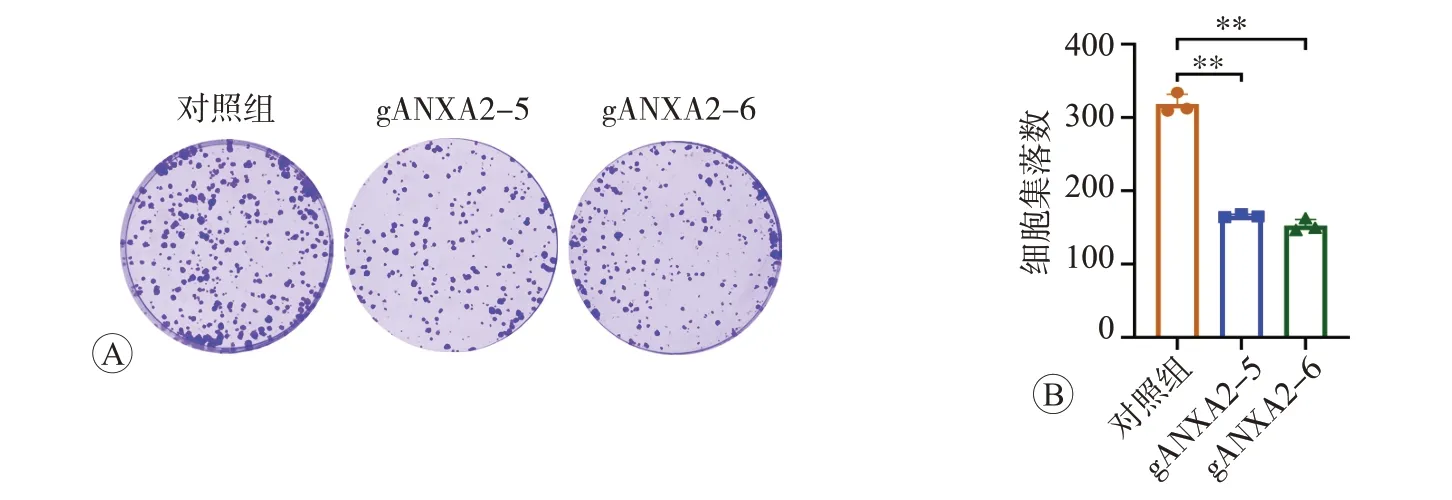
圖6 KYSE30細胞集落形成圖
3 討 論
ANXA2蛋白在人體細胞內廣泛存在,具有多種生物學功能。ANXA2 異常表達參與惡性腫瘤細胞的侵襲、轉移、耐藥等多種過程,其在調控惡性腫瘤進展的關鍵信號通路中發揮重要作用[4]。越來越多的研究表明,ANXA2可能成為新的預后判斷標志物和治療靶點[13]。近年來,基于ANXA2的小分子抑制劑、靶向療法和免疫療法陸續被報道[14-16],其在臨床治療中的應用價值尚待進一步深入探索。
研究表明,ANXA2高表達可以作為食管鱗癌的不良預后標志物[5],其表達升高與食管鱗癌細胞的惡性表型相關,可以通過多種信號通路促進食管鱗癌細胞的侵襲運動[5,17]。這提示ANXA2的異常變化在腫瘤臨床中具有潛在應用價值。
CRISPR/Cas9 是一種高效、簡單、廉價的實現細胞內基因編輯方法。該技術使基因功能和蛋白質功能研究變得更加便捷,基于CRISPR/Cas9 基因編輯技術的疾病治療方法也取得了飛速進展[18]。近些年,研究者們從Cas9蛋白、向導RNA、轉染方法等方面進行了改進,并降低了脫靶效應[19]。以往關于ANXA2基因在腫瘤中的作用研究絕大多數是使用RNA 干擾技術(包括siRNA 和shRNA)進行的。關于ANXA2 敲除的研究目前尚少,有學者使用同源重組技術在人結直腸癌細胞中敲除ANXA2 后,發現細胞的生長和運動均減緩[20];利用CRISPR/Cas9 技術敲除中國倉鼠卵巢細胞中的ANXA2,可以減少重組治療蛋白生產過程中宿主細胞蛋白的污染[21]。然而,目前尚無使用CRISPR/Cas9 技術敲除食管鱗癌細胞中ANXA2基因的研究報道。因此,針對ANXA2 的基因編輯和潛在治療應用有待探索。
本研究使用CRISPR/Cas9 基因編輯技術快速高效地構建了ANXA2 穩定敲低的慢病毒包裝載體,并感染細胞獲得ANXA2基因穩定敲低的食管鱗癌細胞。我們篩選了6 個基因敲低靶點,利用Western blot 和RT-qPCR 實驗檢測基因敲低后細胞內ANXA2 的蛋白和mRNA 水平的變化,最終確定兩個敲除效果較好的靶點進行后續實驗,結果發現ANXA2 敲除細胞中癌基因MYC蛋白表達降低,且細胞的遷移、侵襲以及集落形成能力均明顯減弱。以上結果與本課題組之前報道的使用siRNA 瞬時敲降處理對細胞表型和下游分子表達的影響均一致[5]。我們還觀察到,在ANXA2表達降低最顯著的細胞中,仍能檢測到少量ANXA2 的表達。推測可能的原因為:在病毒感染細胞的過程中,并非所有細胞的感染效率均一致,且整合于基因組的效率也可能不同,從而導致不同細胞中ANXA2 表達量的差異。后續研究將進一步對上述細胞分離單克隆,以獲得敲低效果更好的細胞,同時還將構建敲低ANXA2的其他食管鱗癌細胞模型。
綜上所述,本研究使用CRISPR/Cas9 構建了穩定敲低ANXA2基因的食管鱗癌KYSE30 和KYSE150 細胞,顯著抑制了細胞的惡性表型,為進一步闡明ANXA2 促進食管鱗癌細胞惡性的作用機制以及基于ANXA2通路新靶點的研究奠定了實驗基礎。
