誤診為CIDP的神經元核內包涵體病1例報告
2023-12-18 04:55:30賴有連黃志珍羅聰麗
中風與神經疾病雜志 2023年11期
關鍵詞:癥狀
賴有連, 黃志珍, 羅聰麗, 林 攀
神經元核內包涵體病(neuronal intranuclear inclusion disease,NIID)是一種罕見的、進展緩慢的神經系統退行性疾病,臨床常表現肢體運動感覺異常、意識障礙、癲癇樣發作、智能減退、共濟失調、周圍神經病變、自主神經功能障礙等,常誤診為其他神經系統疾病,其病理學特征是在中樞神經系統、周圍神經系統以及內臟器官的細胞中存在嗜酸性玻璃樣核內包涵體[1]。既往診斷多依賴于尸檢。隨著日本學者Sone 等人[2]提出的可由皮膚活檢確診此病,本病報道數量增加。經過福建省龍巖市第二醫院倫理委員會的審查同意,現將通過皮膚活檢病理和基因檢測確診的1例NIID患者的臨床診療經過進行報道。
1 病例資料
患者,男,46 歲,以“反復雙下肢無力8年,再發1月余”為主訴于2017年10月9日入院。8年前開始出現雙下肢無力,表現為雙下肢站立、步態不穩,伴有全頭部脹痛,出現反復惡心嘔吐,伴有勃起功能障礙,曾反復出現一過性意識不清,就診于外院完善腰穿檢查后考慮慢性炎性脫髓鞘性多發性神經根神經病(chronic inflammatory demyelinating polyradiculoneuropathy,CIDP)[3],治療后癥狀有好轉(具體治療不詳),但上述癥狀仍反復再發并逐漸加重。既往素健,否認藥物濫用史、中毒史及家族性遺傳病史。查體:體溫36.9 ℃,脈搏80 次/min,呼吸20 次/min;血壓130/90 mmHg,心肺腹查體未及明顯異常。專科檢查:神志清楚、對答切題,計算力及定向力正常,雙側瞳孔等圓等大、對光反射靈敏,眼球運動正常、未見水平及垂直眼震,額紋對稱、雙側鼻唇溝對稱,伸舌居中、懸雍垂居中、雙側咽反射弱,四肢肌張力減低,雙上肢肌力5 級、雙下肢肌力4 級,四肢腱反射弱,四肢末端肌肉萎縮,深淺感覺雙側對稱正常,雙側病理征陰性,腦膜刺激征陰性,共濟試驗正常。蒙特利爾認知評估量表(Montreal Cognitive Assessment,MoCA):22分。輔助檢查:血常規、肝腎功能電解質、乙肝二對半、肌酸激酶及同工酶、凝血功能、血脂、甲狀腺功能、血氨、血乳酸、梅毒螺旋體抗體測定、人類免疫缺陷病毒抗體、免疫結締組織病全套、血管炎全套、腫瘤標志物全套等常規實驗室檢查均未見異常。入院診斷:雙下肢無力待查,CIDP可能。肌電圖提示:左側正中神經末梢潛伏期稍延長4.4 ms,雙側正中神經運動傳導速度均減慢;雙側尺神經末端潛伏期延長(左側3.9 ms,右側4.0 ms),復合肌肉動作電位(compound muscle action potential,CMAP)波幅下降;雙側腓總神經CMAP波幅明顯下降、運動傳導速度明顯減慢;雙側脛神經F波出波率下降、最短潛伏期延長;雙側脛神經H反射未引出。肌電圖結果詳見表1。脫髓鞘灶。因受限于當時對NIID 的認知不足,未進一步診斷,治療上予營養神經、改善微循環等處理,癥狀有改善,建議上級醫院進一步診治。
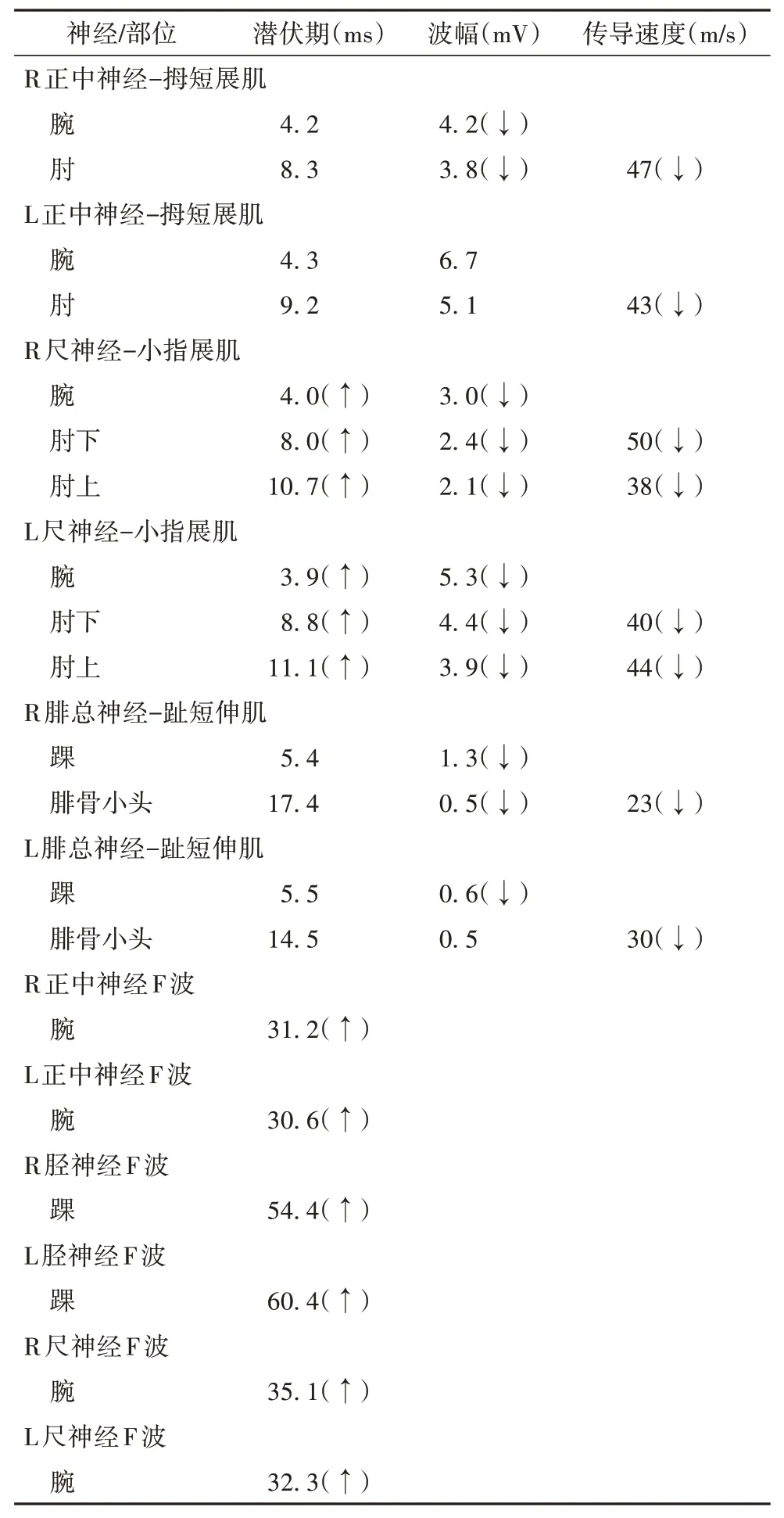
表1 肌電圖結果
2020年9月7日患者因雙下肢無力、反復頭痛、惡心嘔吐再次就診我院,完善頭部彌散加權成像(diffusion weighted imaging,DWI)(見圖1)可見雙側大腦皮髓質交界區呈彌散受限高信號病灶,考慮NIID可能,取得患者及其家屬同意,完善皮膚活檢病理檢查和NLCGGC相關疾病譜基因檢測,遂于左側踝關節上10 cm 處取皮膚活檢送首都醫科大學附屬北京天壇醫院行病理檢查,免疫組織化學染色結果提示:部分汗腺細胞、脂肪細胞和纖維細胞的細胞核內可見P62、泛素抗體強陽性染色的包涵體(見圖2)。診斷NIID明確。
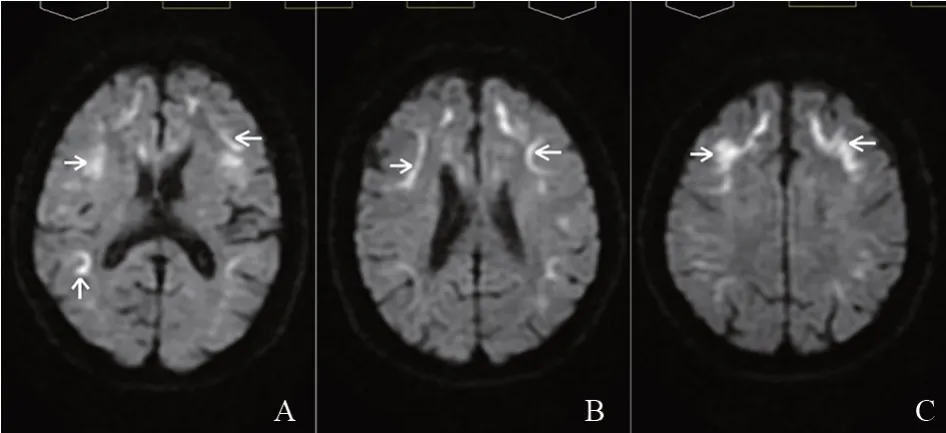
圖1 頭部核磁共振圖像
腰穿腦脊液檢查:壓力100 mmH2O;常規、生化、免疫、三大染色及細胞學結果均未見明顯異常。外院查頭部磁共振平掃示雙側額顳頂枕皮質下對稱性該患者基因檢測結果(見圖3)提示NOTCH2 NLC基因中GGC異常擴增次數為142次。
2 討 論
NIID 是一種罕見的慢性進展性神經變性疾病,根據發病的年齡可分為嬰兒型、青少年型和成人型,Sone等[1]據家族發病情況將NIID分為家族型及散發型,又據早期臨床表現分為癡呆型和肢體無力型。典型成人型NIID 包括認知障礙和白質腦病,臨床表現無明顯特異性。一般來說,散發型NIID 發病年齡較大,家族型NIID 發病年齡較小,且肢體無力為首發癥狀較多[4]。楊帆等人[5]將NIID 主要癥狀歸納為3 個類型:(1)中樞神經系統受累,出現癡呆、共濟失調、發作性意識障礙、行為異常、亞急性腦炎樣表現、強直、震顫、癲癇發作、卒中樣發作等;(2)周圍神經受累,出現感覺障礙、遠端肌力下降;(3)自主神經受累,表現為瞳孔縮小、尿失禁、嘔吐、暈厥等。本例患者以反復進行性雙下肢無力癥狀起病,病程進展過程中出現周圍神經損害、性功能障礙、反復發作性意識障礙、輕度認知功能障礙等表現,且家屬無相關癥狀,符合散發型NIID 表現。但該患者癥狀進展超過8周,呈慢性進展病程,表現為對稱性雙下肢無力,四肢腱反射減弱,外院診斷為CIDP,我院肌電圖提示周圍神經傳導速度減慢、傳導阻滯,無明顯中毒、遺傳、腫瘤、自身免疫疾病等其他周圍神經病的證據,也支持CIDP 的肌電圖表現[6],導致該病例極易誤診。因此,NIID的臨床表現具有很強的異質性。
NIID的特征性影像學表現為DWI上皮髓質交界處(U型纖維)出現有高信號,這種征象隨著疾病進展,病灶將不斷地沿皮質向后延伸,不會向髓質深部延伸,陳為安等人[7]將其命名為“皮質下綢帶征”。該患者的典型DWI特征為我們確診NIID提供了關鍵線索。
進一步確診NIID 主要依靠病理活檢,在2011年Sone 等人[8]報道了在皮膚活檢的標本中發現了脂肪細胞、成纖維細胞和汗腺細胞內可見嗜酸性核內包涵體,這些核內包涵體泛素化陽性。2019年,唐北沙教授團隊[9]通過長讀長測序(longreadse-quencing,LRS)首次揭示了NIID 致病機制與NOTCH2NLC基因中GGC異常重復擴增相關,且NIID臨床表現的多樣性可能與NOTCH2NLC基因5'區域GGC病理性重復次數相關,一般認為GGC重復擴增次數超過60次具有致病性。
綜上所述,NIID臨床表現多樣,其病理表現為神經元核內出現嗜酸性包涵體,影像學可表現為經典“綢帶征”,癥狀上可與多種神經變性病相疊加,其發病與NOTCH2NLC基因5'區域GGC 病理性重復次數相關。本例NIID 如無影像學檢查作為線索,將一直被認為是CIDP,這提示我們在臨床診療中發現周圍神經病變的患者,也應當重視和排查中樞神經系統是否累及。因此,呼吁我國NIID 協作組開展針對NIID的研究,深入了解其發病機制,并不斷完善制定相關診斷標準和流程。
倫理學聲明:本研究方案經由福建省龍巖市第二醫院倫理委員會審批(批號:LYEYEC 2022-005),患者已簽署知情同意書。
利益沖突聲明:所有作者均聲明不存在利益沖突。
作者貢獻聲明:賴有連、林攀負責論文設計;賴有連負責繪制圖表和撰寫論文;賴有連、羅聰麗、黃志珍、林攀負責數據及文獻收集;林攀負責擬定寫作思路、指導撰寫文章并最后定稿。
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
