LC-ESI-MS/MS測定白酒中氨基甲酸乙酯的方法研究
2023-12-19 12:43:16董孝元汪明迪唐艷榮
釀酒科技 2023年11期
董孝元,周 玉,余 義,劉 莎,汪明迪,孫 莉,唐艷榮*
(1.黃鶴樓酒業有限公司,湖北 武漢 430050;2.湖北信嘉檢測科技有限公司,湖北咸寧 437000;3.黃鶴樓酒業(咸寧)有限公司,湖北咸寧 437000)
中國是蒸餾酒生產大國。酒類在發酵蒸餾過程中會產生一種致癌物質氨基甲酸乙酯,該物質不斷為大眾所熟知,是食物在發酵或貯存過程中天然產生的物質,普遍存在于發酵食品和酒類產品中。眾所周知,用糧食釀造的白酒需要經過發酵以及高溫蒸煮,釀造酒中的氨基甲酸乙酯主要是由乙醇和尿素共同作用產生的。尿素是在發酵過程中酵母分解精氨酸所產生的副產品。氨基甲酸乙酯也會在蒸餾酒內產生,特別是核果(如櫻桃、杏和梅)烈酒。果核所含的氰基糖苷在酶水解后會產生一種名為異氰酸酯的副產品,異氰酸酯與酒中的乙醇發生化學反應產生氨基甲酸乙酯。
氨基甲酸乙酯是一種廣泛存在于白酒飲料中的2A 類致癌物質,對人體健康存在潛在危害性[1],各類酒中均不同程度微量存在,一些國家對酒中的含量有明確的限制規定[4]。關于食物中的EC含量,國際上對食品及酒類的氨基甲酸乙酯含量控制規定各不相同。我國白酒固態發酵方式及獨特的生產工藝,在一定程度上限制了現有氨基甲酸乙酯控制措施的可使用性。目前氨基甲酸乙酯的檢測方法包括液相法、液相質譜串聯法、氣相質譜串聯法等[3]。本文從利用液相質譜聯用儀高效快速檢測氨基甲酸乙酯入手,優化前處理方式,縮短檢測出峰時間,有利于我國白酒中氨基甲酸乙酯限量標準的建立,為消費者和白酒生產企業提供參考,使我國白酒企業得以良性發展及保障白酒產品的質量安全[2]。
1 材料與方法
1.1 材料、試劑及儀器
樣品:市售白酒。
試劑及耗材:氨基甲酸乙酯(規格0.1 g,儲存溫度20 ℃±4 ℃),Dr.Ehrenstorfer GmbH;乙腈(色譜純),美國Baker 公司;甲酸(色譜純),麥克林集團有限公司;超純水,德國默克密理博MiLLi-QDireat。
儀器設備:Agilent 1260LC-Agilent 6460 安捷倫三重串聯四級桿質譜儀;Agilent ZORBAX RXC18安捷倫色譜柱(2.1×150 mm,1.8 μm);有機濾膜(孔徑0.22 μm),天津津騰;天津津騰全玻璃微孔過濾器(規格1000 mL)。
1.2 實驗方法
1.2.1 標準溶液配制
標準儲備液(100 mg/L):準確稱取10 mg(精確至0.0001 mg)氨基甲酸乙酯標準品于100 mL 容量瓶中,用一級水溶解并定容至100 mL,配制成100 mg/L的標準儲備液,置于4 ℃冰箱中保存。
標準工作液:分別準確吸取0.04 mL、0.08 mL、0.16 mL、0.40 mL、0.80 mL 標準儲備液于100 mL容量瓶中,一級水定容至刻度,配制成濃度分別為0.04 mg/L、0.08 mg/L、0.16 mg/L、0.40 mg/L、0.80 mg/L的標準工作液。
1.2.2 樣品前處理方法
用移液管準確移取10 mL 白酒樣品于蒸發皿中,放于沸騰水浴鍋上,蒸發至近干后用純水定容至10 mL,并搖勻。用一次性2.5 mL 注射器吸取均質樣品,經0.22 μm的有機濾膜過濾至1 mL進樣瓶中,供液相色譜-質譜聯用儀上機檢測。
1.2.3 色譜條件
(a)色譜柱:C18柱(柱長150 mm,柱內徑2.1 mm,填料粒徑1.8μm);
(b)流動相:A 相:0.1 %甲酸溶液;B 相:乙腈溶液;
(c)流速:0.15 mL/min;
(d)柱溫:40 ℃;
(e)進樣量:2 μL。
1.2.4 質譜條件
(a)離子源類型:電噴霧離子源;
(b)掃描方式:正離子模式;
(c)毛細管電壓:3000 V;
(d)離子源溫度:350 ℃;
(e)霧化氣:30 psi;
(f)多反應監測:選擇至少2個子離子。
2 結果與分析
2.1 色譜條件和質譜條件的優化
2.1.1 質譜條件的優化
將液相部分色譜柱換為兩通連接,采取直接進樣的方式,將配制的純品目標物標準溶液注入質譜,分別進行正離子模式和負離子模式,在Scan 狀態下進行全掃描檢測,得到TIC 總離子流圖,根據總離子流圖標記出特征離子峰,即得到一級質譜圖和準分子離子峰,再進行SIM 模式掃描,將標記出的準分子離子峰進行單獨掃描,得到EIC 提取離子流圖;再用碰撞氣惰性氣體氮氣攻擊該前體離子,獲得其二級質譜圖及相應的子離子。結果表明在正離子模式下響應值及靈敏度均高于負離子模式,因此,本方法選擇正離子電離模式。利用多反應監測模式(MRM))對選定的定性離子和定量離子對裂解電壓、毛細管電壓、碰撞能、保留時間、碰撞池加速電壓等質譜參數進行優化,使各監測離子豐度和信號達到最佳。正離子模式下的質譜分析參數見表1。

表1 氨基甲酸乙酯的質譜參數
2.1.2 色譜條件的優化
通過對氨基甲酸乙酯物理和化學信息的分析,分別以0.1%甲酸-甲醇、0.1%甲酸-乙腈、5 mmol/L甲酸銨-乙腈、5 mmol/L 甲酸銨-甲醇作為流動相進行樣品分離。由于不同流動相的pH 值不同,對色譜柱中的填料物質會起到一定的作用,有的流動相有利于目標物質的分離,有的流動相則會抑制目標物出峰,導致峰形異常,如峰拖尾、峰前伸、雙頭峰,以及鬼峰等現象,均不利于對樣品進行定性以及定量分析。通過利用以上流動相對氨基甲酸乙酯進行色譜峰分離,結果表明,用0.1%甲酸-乙腈作為流動相時物質分離度、峰形、響應值以及保留時間的穩定性均優于0.1%甲酸-甲醇、5 mmol/L甲酸銨-乙腈、5 mmol/L 甲酸銨-甲醇,故本方法最終選用0.1 %甲酸-乙腈為流動相進行洗脫。圖3 是氨基甲酸乙酯在優化的色譜和質譜條件下的MRM色譜圖。
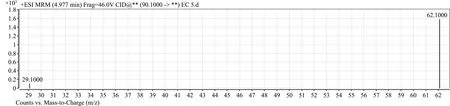
圖1 氨基甲酸乙酯離子質譜圖
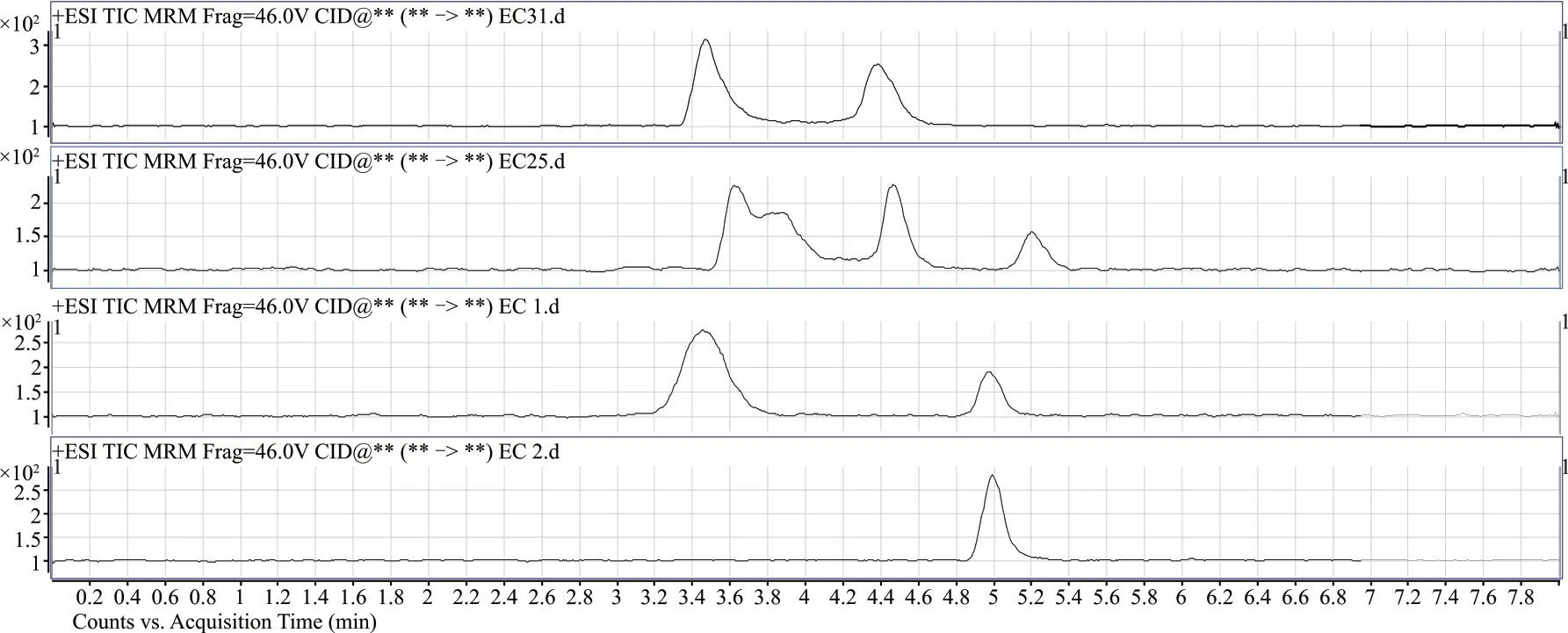
圖2 不同流動相色譜圖對比
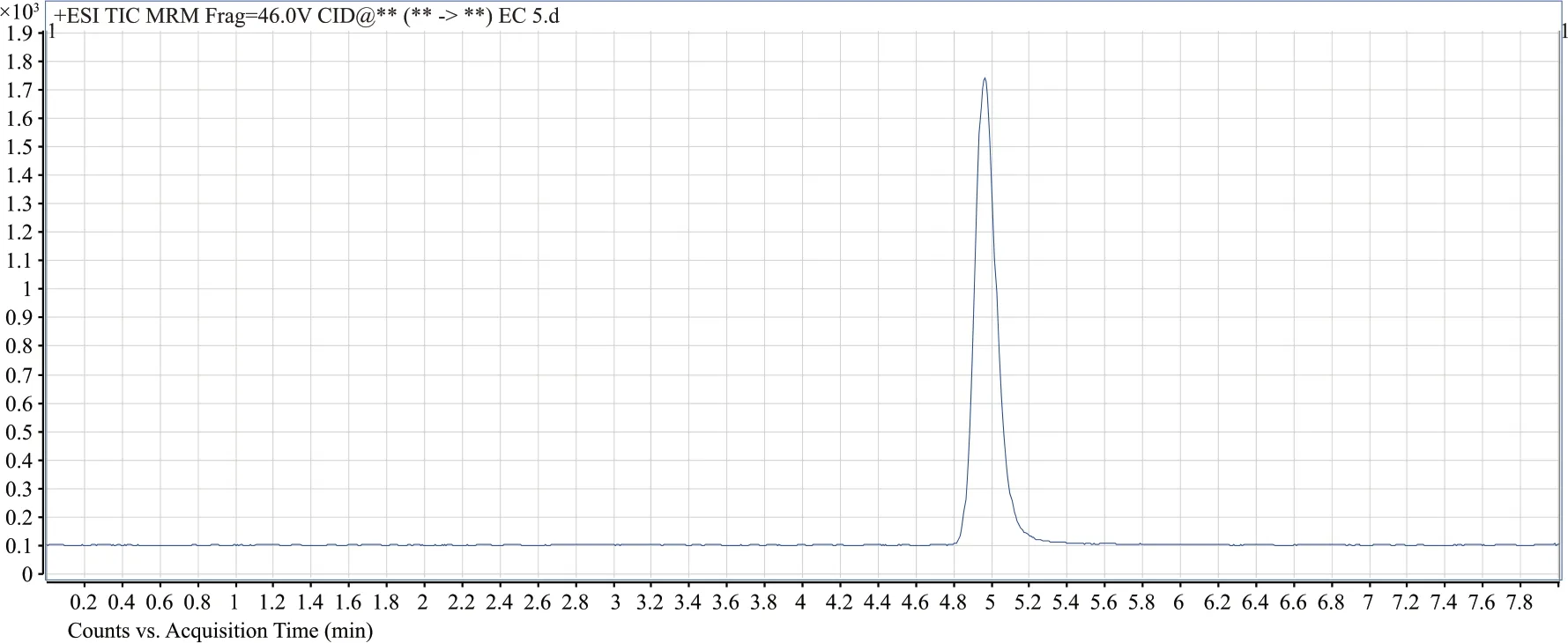
圖3 氨基甲酸乙酯的多反映監測色譜圖
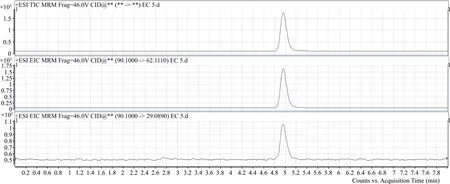
圖4 氨基甲酸乙酯母離子及子離子色譜圖
2.2 線性范圍、檢出限和定量限
將氨基甲酸乙酯標準儲備液用一級水稀釋,分別配制成濃度為40 μg/L、80 μg/L、160 μg/L、400 μg/L、800 μg/L 的標準工作液,按1.2 節所述的色譜-質譜條件進行測定,繪制標準工作曲線,如圖5 所示。參考標準GB/T 27417—2017《合格評定化學分析方法確認和驗證指南》中5.4 檢出限和定量限的評定方法規定,信噪比法評估檢出限和定量限,檢出限可接受的信噪比為3∶1,定量限可接受的信噪比為10∶1。本方法為保證實用性,模擬氨基甲酸乙酯在白酒中的狀態,由于目前市面上的白酒酒精度大多在42%vol和52%vol,本方法利用購買的無水乙醇,配制成酒精度為46%vol 的白酒模擬樣品,在白酒模擬樣品中添加氨基甲酸乙酯標準儲備液,進行方法探底試驗。既要保證信噪比可接受,也要保證峰形易識別,且峰形無異常,如圖6所示。
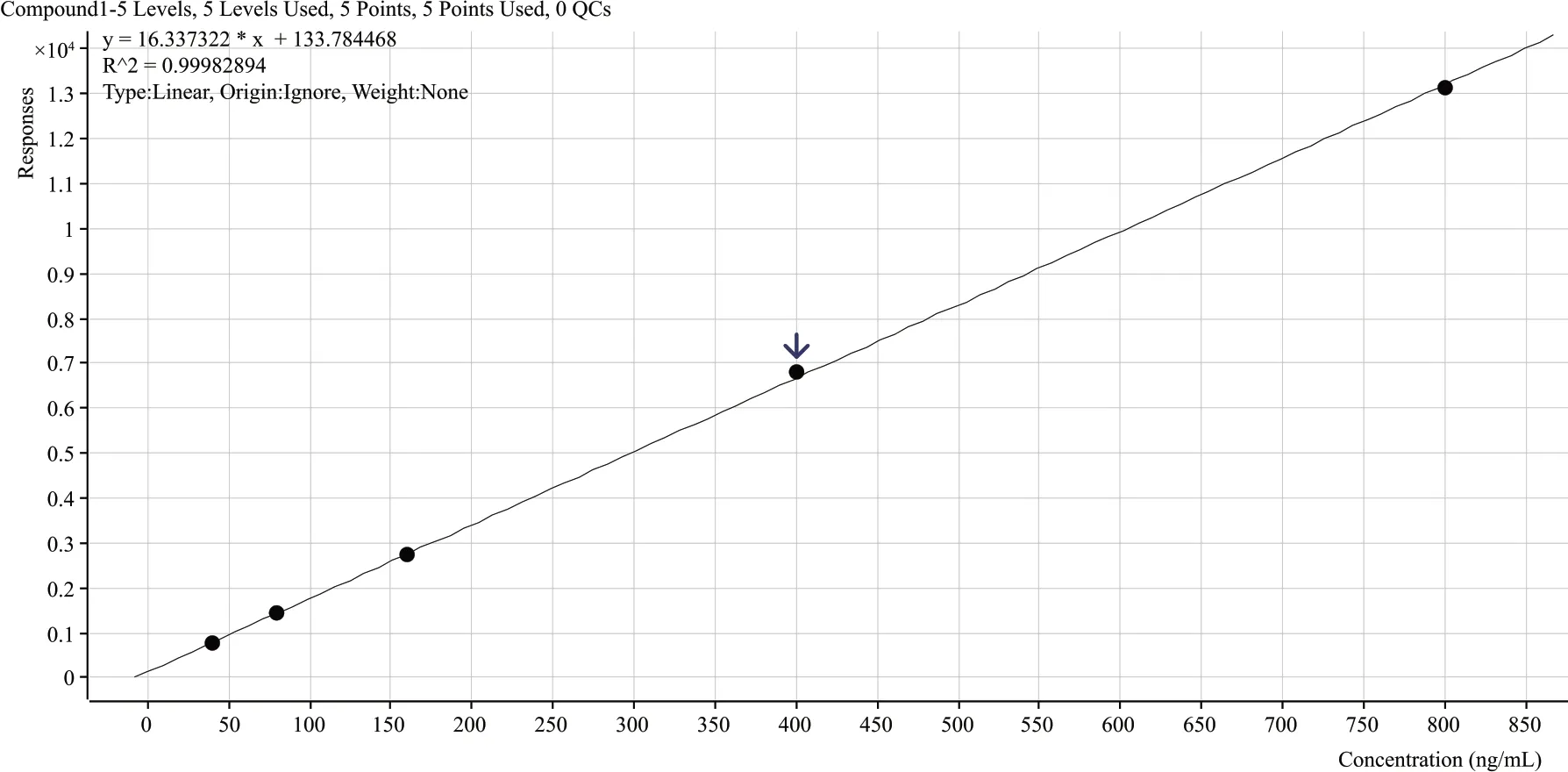
圖5 氨基甲酸乙酯的線性相關系數

圖6 檢出限和定量限信噪比
結果顯示,在40~800 μg/L 的濃度范圍內,分析物氨基甲酸乙酯的質量濃度x(ng/mL)和響應值y之間線性相關系數為0.999;模擬樣品中添加濃度為10 μg/L 時,信噪比為6 大于3,峰形易識別且無異常,即確定為檢出限;濃度為20 μg/L信噪比為15大于10,峰形易識別且無異常,故確定為定量限。
2.3 精密度和回收率
為充分保證相關樣品檢測數據的準確有效,本試驗分別準備3 種常見的不同香型白酒樣品,濃香型白酒、醬香型白酒和清香型白酒,并在3 種白酒樣品中分別加入按標準工作曲線濃度最高點的0.1倍、0.5倍和0.9倍的標準溶液,配制成低中高3種濃度的加標樣品溶液,按照給定分析方法的全過程進行處理和測定,記錄6 個平行樣的測定結果,按全程序每個樣品平行測定6 次,分別計算每個樣品的加標回收率,統計其相對標準偏差,見表2、表3、表4。
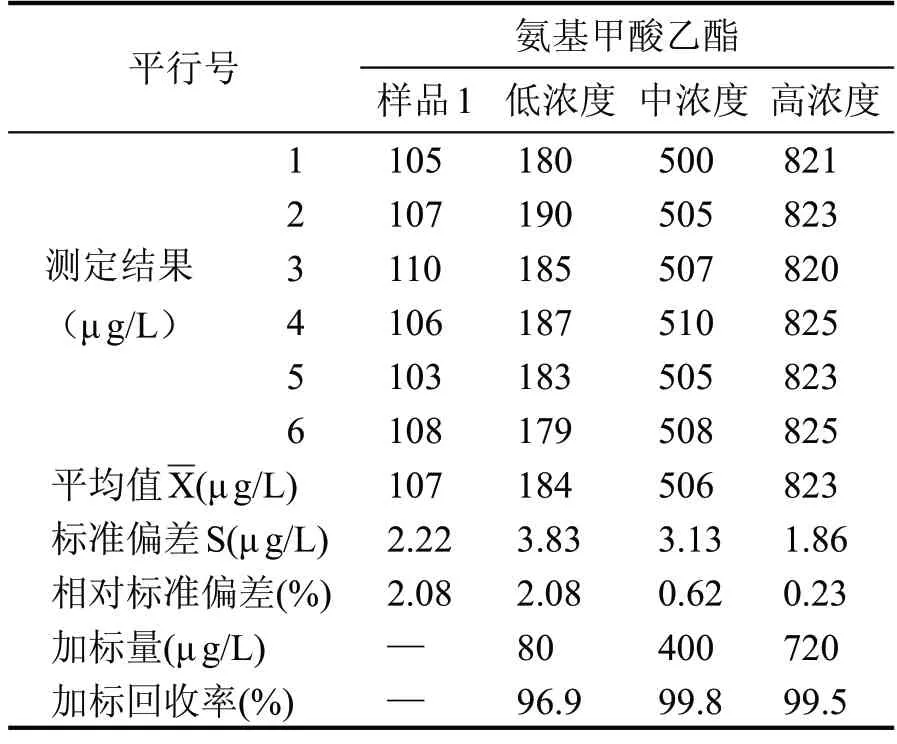
表2 樣品1加標回收率計算
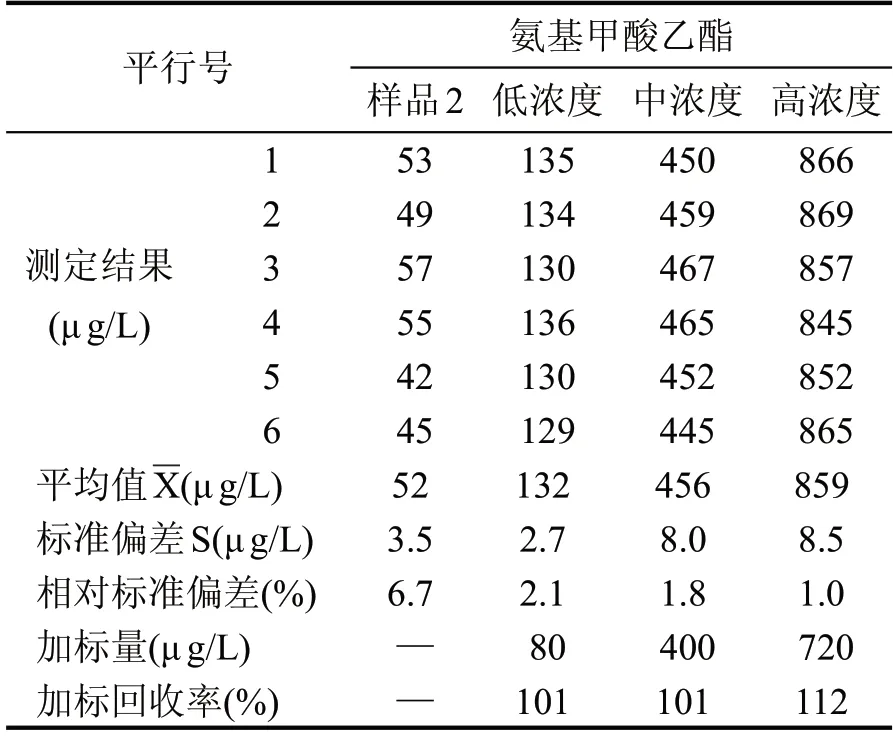
表3 樣品2加標回收率計算
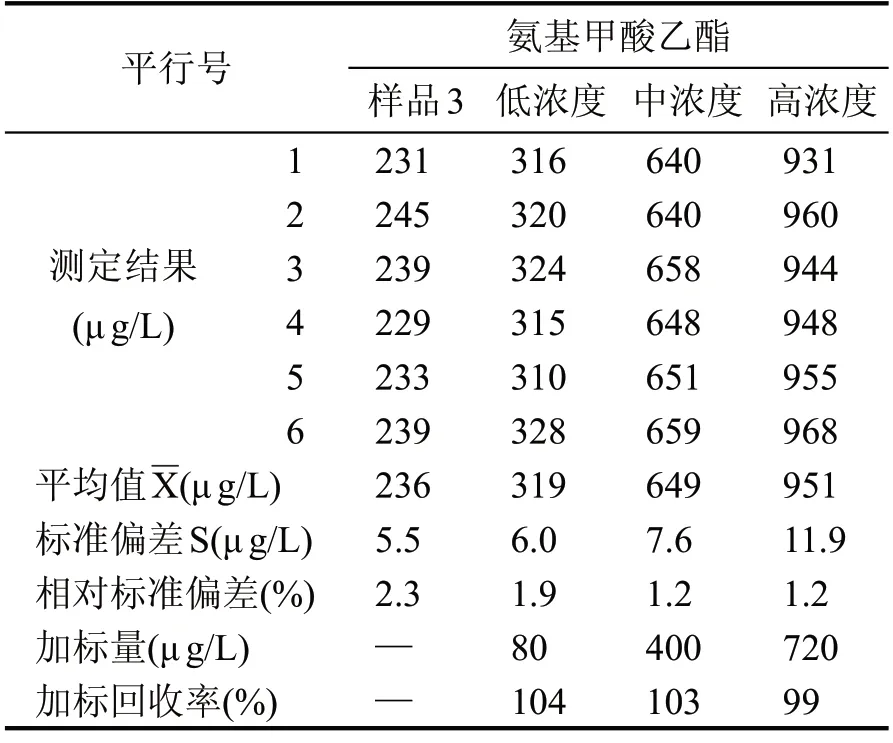
表4 樣品3加標回收率計算
參考公式:
加標回收率=(加標試樣測定值-試樣測定值)/加標量×100%
由表2 可知,本實驗中檢測結果的重復性相對標準偏差(變異系數)均小于10 %,且加標回收率均在95%~115%之間,滿足方法驗證學要求。
2.4 與國標法比對
選擇市面上常見的3 種香型白酒,濃香、清香和醬香,按照本方法和食品安全國家標準食品中氨基甲酸乙酯的測定GB 5009.223—2014 分別進行樣品前處理以及上機檢測。本方法采用除醇方式對白酒樣品進行前處理,液相色譜-質譜聯用儀進行檢測,外標法定量分析;國標GB 5009.223—2014采用萃取、分液及氮吹等方式對樣品進行前處理,氣相色譜-質譜儀進行檢測,內標法定量分析。3種香型白酒的檢測比對結果見表5。
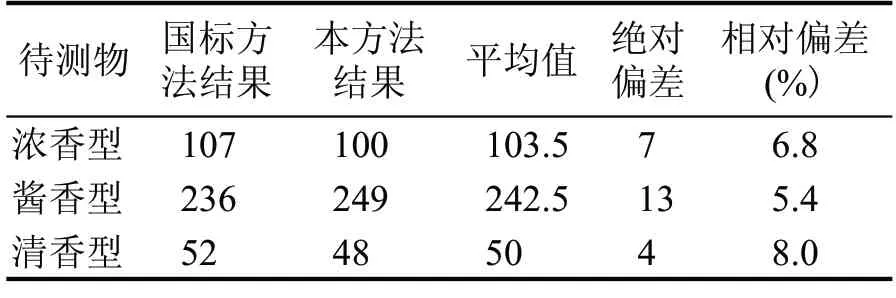
表5 方法結果比對 (μg/L)
由表5 可知,不同香型白酒氨基甲酸乙酯含量差別比較大,將本方法和國標方法的數據進行比較,相對偏差均小于10 %,由此可知,對于不同濃度的白酒樣品,本方法與國標方法檢測結果相差不大,能滿足檢測要求。
3 結論
本研究利用液相色譜-質譜聯用儀分析了白酒中氨基甲酸乙酯化合物的分子信息,利用多反應監測模式的液相色譜-質譜聯用技術,建立了快速、穩定、靈敏的可定性定量分析氨基甲酸乙酯的方法。該方法可在8 min內完成,經過多種方法學驗證,其檢測范圍內線性良好(r=0.999);檢出限0.01 mg/L;定量限0.02 mg/L;精密度小于10 %;回收率大于95%;并且在與國標方法檢測結果比較中,檢測結果相對偏差均小于10 %。對于不同香型白酒,氨基甲酸乙酯濃度各異。由結果可知本方法與國標方法偏差較小,滿足使用要求。與國標中的檢測方法相比本方法前處理更加方便快捷,便于日常對白酒中氨基甲酸乙酯的含量進行監控。綜上所述本方法可以對白酒中氨基甲酸乙酯進行實際的定性定量分析檢測。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
