單基因高血壓的研究進展
2024-04-29 17:32:49張玉帕麗達阿布來提
心血管病學進展 2024年2期
張玉 帕麗達?阿布來提

【摘要】單基因高血壓是一種重要的高血壓遺傳模式,它是一種由遵循孟德爾遺傳的單一遺傳變異引起的。目前,單基因高血壓的臨床發病率及發病機制尚不完全清楚,臨床不易識別與診斷。現旨在對單基因高血壓的分類、突變基因及機制特點、診斷治療等方面進行系統闡述,以期為今后該疾病在臨床的診療提供更多的參考。
【關鍵詞】單基因高血壓;孟德爾遺傳;基因檢測
【DOI】10.16806/j.cnki.issn.1004-3934.2024.02.000
Monogenic Hypertension
ZHANG?Yu,Palida·Abulaiti
(Third departments of comprehensive internal medicine,The First Affiliated Hospital of Xinjiang Medical University,Urumqi 830000,Xinjiang,China)
【Abstract】Monogenic hypertension is an important genetic pattern of hypertension,which is caused by a single genetic variant that follows Mendelian inheritance. At present,the clinical incidence and pathogenesis of monogenic hypertension are not completely clear,and it is difficult to identify and diagnose clinically. The purpose of this paper is to systematically elaborate the classification,mutated genes,mechanism characteristics,diagnosis and treatment of monogenic hypertension,in order to provide more references for the clinical diagnosis and treatment of this disease in the future.
【Keywords】Monogenic hypertension;Mendelian inheritance;Genetic testing
高血壓是全球心血管疾病罹患和過早死亡的主要原因之一,目前高血壓的患病率仍呈上升趨勢,而且由于其長期控制不佳,會造成多種靶器官損害,死亡率也日益增高[1]。單基因高血壓(monogenic hypertension,MH)是由單一基因突變引起的高血壓病,是高血壓病的一種罕見類型。它的主要特征為早發嚴重高血壓、酸堿代謝紊亂(最常見的是堿中毒和低鉀血癥)、低腎素水平以及陽性家族史[2]。其患病率不詳,臨床不易辨識與診治,近年來,隨著對MH發病機制的深入研究,對其突變基因的探究已成為新的熱點,現簡述MH的臨床分類,重點綜述MH在已知突變基因、診斷和治療方面的進展。
1 ?MH的常見分類方法
MH的遺傳形式源于鹽皮質激素、糖皮質激素或交感神經通路中的功能獲得或喪失突變。因此,根據突變基因的不同表型,MH有不同的臨床分類方法。當突變引起鹽皮質激素的功能改變時,我們常根據血清腎素和醛固酮水平對MH進行分類。首先,根據血清腎素水平可將其分為正常腎素MH和低腎素MH,而低腎素MH可根據醛固酮水平進一步分類:低醛固酮低腎素性高血壓、正常醛固酮低腎素性高血壓或高醛固酮低腎素性高血壓。另一類是由于腎上腺素/交感神經功能突變引起的MH[3]。根據鹽敏感性和醛固酮水平,MH合并低鉀血癥可分為三類:鹽不敏感型高血壓、鹽敏感低醛固酮型高血壓、鹽敏感高醛固酮型高血壓[2]。
同時,在臨床上,MH也會用到其他的分類方法。腎臟和腎上腺是參與MH的兩個主要器官,根據兩個器官上的基因突變可分為:腎上腺突變型高血壓和腎遠曲小管、集合管突變型高血壓[4]。另外,MH根據病理生理機制的不同可以分為五類:(1)醛固酮合成過度;(2)腎上腺類固醇代謝和作用失調;(3)遠端小管中鈉和氯轉運體的過度活動;(4)腎上腺素能過剩;(5)血管平滑肌增生[5]。不論其如何分類,所有的MH都具有高血壓這一共同臨床表現。
2 ?MH的常見亞型
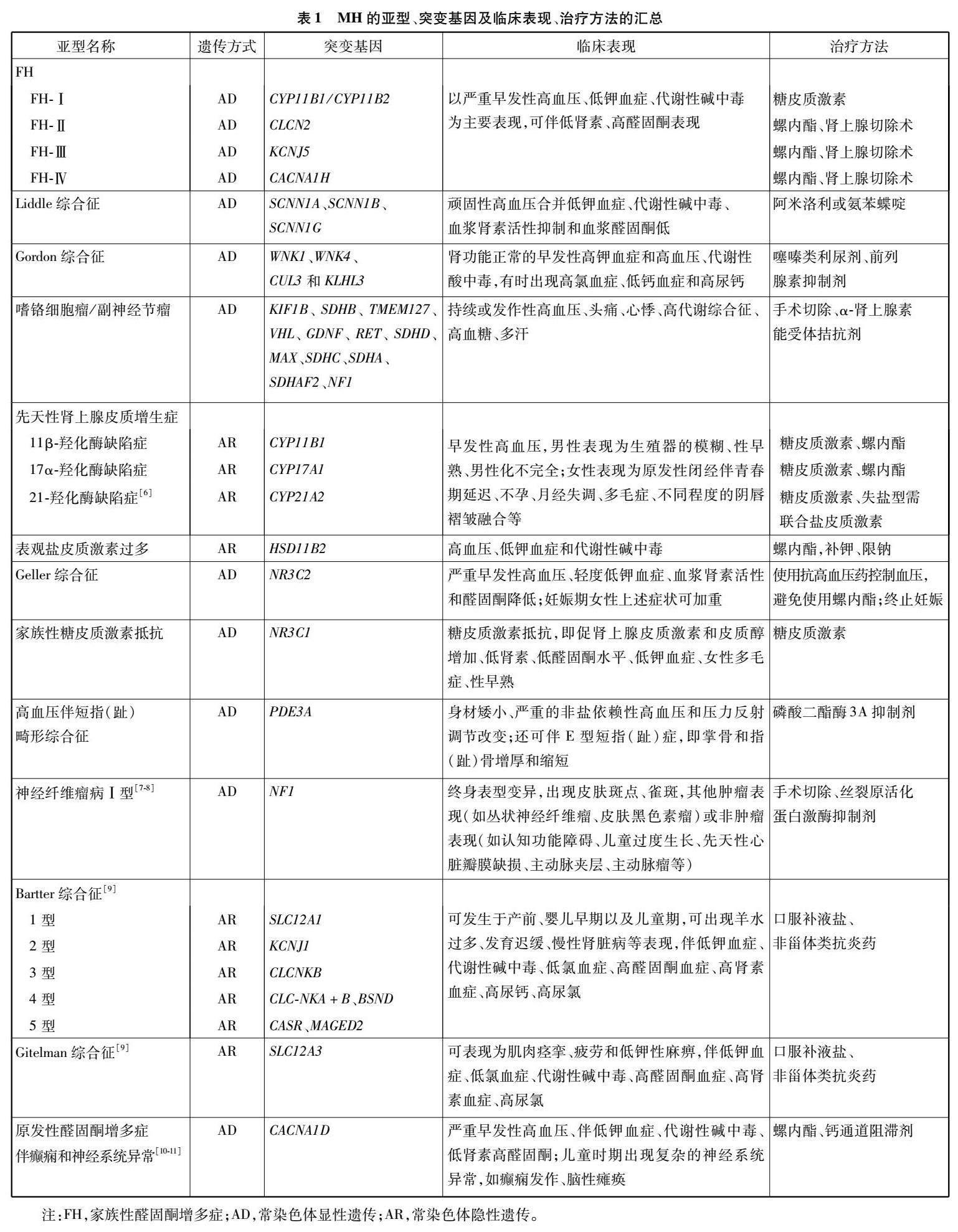
目前,已明確突變基因的MH亞型有22種(見表1)。在這里,我們詳述了MH幾種常見亞型及其遺傳方式、突變基因、臨床表現、治療方法
2.1??家族性醛固酮增多癥
家族性醛固酮增多癥(familial hyperaldosteronism,FH)是一種罕見的常染色體顯性遺傳(autosomal dominant,AD)的MH。它是一組以嚴重早發性高血壓、低鉀血癥、代謝性堿中毒和血漿醛固酮/腎素濃度比值(aldosterone to renin ratio,ARR)升高(ARR>20)為特征的疾病[12]。在臨床上,FH的各種亞型很難與散發性原發性醛固酮增多癥(primary aldosteronism,PA)區分開來。根據潛在的遺傳缺陷,FH可分為Ⅰ~Ⅳ型。其中,FH-Ⅰ約占PA的0.6%[12],是由11β-羥化酶基因(11 β-hydroxylase,CYP11B1)和醛固酮合成酶基因(aldosterone synthase,CYP11B2)的不對稱交叉產生嵌合基因所致,該基因在CYP11B1的5端具有促腎上腺皮質激素(adrenocortical hormone,ACTH)反應啟動子區,在CYP11B2的3端具有醛固酮合成酶編碼區,導致醛固酮的產生依賴于ACTH的表達[13],由于糖皮質激素對該疾病有顯著的積極作用,它也被命名為糖皮質激素可治性醛固酮增多癥(glucocorticoid-remediable?aldosteronism,GRA)。FH-Ⅱ在PA患者中的患病率為1.5%~6.0%[12],它是由CLCN2基因突變所致,CLCN2編碼腎上腺腎小球中表達的電壓門控氯離子通道2?型,該基因突變使腎小球細胞膜容易去極化并激活電壓門控鈣通道,上調編碼醛固酮合成酶的CYP11B2的表達,從而使鹽皮質激素通道過度激活[14]。FH-Ⅲ約占PA的0.3%[15],可能是由KCNJ5的突變引起的,KCNJ5編碼G蛋白偶聯內向整流鉀通道4(G-protein-coupled?inward?rectifier?K+?channels?4,?GIRK4)該基因突變影響GIRK4的選擇性,導致腎上腺皮質細胞中鉀選擇性的喪失,增強鈉電導,增加鈉的流入和膜的去極化,最終導致CYP11B2的表達升高[16]。FH-Ⅳ是由CACNA1H突變引起[17],在全球范圍內的發病率尚不清楚。CACNA1H在腎上腺球狀帶中大量表達,并編碼T型鈣通道的α亞基(CaV3.2)。CaV3.2的異常激活,增加鈣內流,引起去極化,導致醛固酮合成異常[18]。使用低劑量的皮質類固醇可以有效抑制GRA患者ACTH的分泌,進而降低醛固酮水平。而FH-Ⅱ、FH-Ⅲ、FH-Ⅳ均需要通過單側腎上腺切除術聯合鹽皮質激素拮抗劑來緩解癥狀[19]。
2,2??Liddle綜合征
Liddle綜合征(liddle syndrome,LS)是一種常染色體顯性遺傳疾病,是由編碼上皮鈉通道(epithelial sodium channel,ENaC)a、b、c亞基的SCNN1A、SCNN1B和SCNN1G基因突變引起的。遠端小管中過度活化的ENaC直接導致鈉重吸收增加,繼而導致血容量擴張[10,19]。它可能是MH最常見的形式之一。有研究[20]發現在中國年輕高血壓患者中,LS發病率為0.91%~1.52%。然而,它在整個高血壓人群中的患病率仍然未知。LS最具特征的表現是頑固性高血壓合并低鉀血癥、代謝性堿中毒、血漿腎素活性抑制和血漿醛固酮低,通常與猝死和早發性高血壓的陽性家族史有關。截至目前,全球共發現41種導致LS的突變體,其中22種由中國學者首次報道[19]。在LS的治療中,首選ENaC阻斷劑(阿米洛利和氨苯蝶啶),并輔助低鈉飲食。在中國阜外醫院的國家心血管疾病中心建立的世界上最大的LS隨訪隊列中,共有來自13個家系的74名患者接受了阿米洛利治療并定期隨訪,治療有效率為90%[5,19]。
2.3 ?Gordon綜合征
Gordon綜合征(gordon syndrome,GS),又稱2型假性低醛固酮減少癥(pseudohypoaldosteronism type II,PHAⅡ),是由調節腎遠曲小管Na-Cl協同轉運蛋白(NaCl cotransporter,NCC)活性的WNK1、WNK4、CUL3和KLHL3的功能獲得突變引起的,表現為腎功能正常的早發性高鉀血癥和高血壓、代謝性酸中毒,有時出現高氯血癥、低鈣血癥和高尿鈣[10,19]。高鉀血癥是GS的一個特征,其值可能為9 mmol/L。然而,由于異質性突變,患者可以表現出正常的血鉀水平。在治療中,噻嗪類藥物對NCC具有抑制作用,因此臨床建議使用噻嗪類利尿劑,有助于改善電解質異常并使血壓正常化。對于輕度變異的患者,單獨限制飲食中的鹽可能是有效的[5,19]。
2.4 ?嗜鉻細胞瘤/神經節旁瘤
嗜鉻細胞瘤/神經節旁瘤(pheochromocytoma/paraganglioma,PCC/PGL)也是MH的常見病因之一,在高血壓患者中的患病率為0.1%~0.6%[21]。遺傳性PCC可單獨發生或作為某些遺傳性腫瘤綜合征的局部表現,如多發性內分泌腫瘤、神經纖維瘤病等[22]。攜帶易感基因的患者很有可能在年輕時發展為PCC/PGL[23]。迄今為止,已經報道了以下PCC/PGL易感基因:KIF1B、SDHB、TMEM127、VHL、GDNF、RET、SDHD、MAX、SDHC、SDHA、SDHAF2、NF1[24]。這些基因突變激活兒茶酚胺/交感神經系統導致兒茶酚胺的過度釋放、濃度升高,在臨床上可表現為持續或發作性高血壓、頭痛、心悸、高代謝狀態、高血糖和多汗。在明確診斷后,功能性PCC通常通過手術切除,并在圍手術期服用α腎上腺素能拮抗劑[19,25-26]。
2.5 ?先天性腎上腺皮質增生癥
先天性腎上腺皮質增生癥(congenital adrenal hyperplasia,CAH)是一組CYP11B1和CYP17A1突變與糖皮質激素合成有關的疾病,包括11β-羥化酶缺乏癥(11β-hydroxylase deficiency,11β-OHD)——Ⅳ型和17α-羥化酶缺乏癥——Ⅴ型。該類突變導致具有鹽皮質激素活性的腎上腺類固醇如脫氧皮質酮(deoxycorticosterone,DOC)的積聚,從而出現高血壓。11 β-OHD占所有CAH的0,2%~8.0%[19]。由于這種疾病往往伴隨著高雄激素血癥,它會促進體細胞的快速生長和骨骼成熟。此外,還可以觀察到女性的男性化和男性的早熟[10,19]。由于皮質醇合成能力下降和性激素產生受損,在CAH的Ⅴ型中可以觀察到男性的男性化不完全或生殖器模糊,以及女性的原發性閉經伴青春期延遲[27]。CAH患者建議使用糖皮質激素與鹽皮質激素受體(mineralocorticoid receptor,MR)拮抗劑螺內酯聯合抑制ACTH[10,19]。
2.6 ?表觀鹽皮質激素過多
表觀鹽皮質激素過多(apparent mineralocorticoid excess,AME)是一種罕見的常染色體隱性遺傳疾病,其發病率在整個高血壓人群尚不清楚。該疾病由11β羥基類固醇脫氫酶2型(11β-hydroxysteroid dehydrogenase type II,11βHSD2)缺乏引起。在生理狀態下,11βHSD2將皮質醇轉化為可的松,從而保護MR免受皮質醇激活[28]。病理狀態下,MR的激活可誘導高血壓、低鉀血癥和代謝性堿中毒。在50%~75%的患者中,可以觀察到由高尿鈣引起的腎鈣沉著癥,更罕見的是由于慢性低鉀血癥引起的腎囊腫[14]。AME的診斷依靠測量24?h尿游離皮質醇與可的松的比值,正常比值為1:1,而AME患者的比值范圍為6.7~33.0。AME患者建議服用MR拮抗劑螺內酯,輔以低鈉飲食及低劑量地塞米松來抑制促腎上腺皮質激素而引起的高血壓和堿中毒[19,29]。
2.7 ?Geller綜合征
Geller綜合征,又稱妊娠加重高血壓,具有AD遺傳模式,由MR突變引起,該突變存在于染色體4q31上,在密碼子810(S810L;MRL810)處亮氨酸取代絲氨酸。這一突變改變ENaC和鈉鉀泵結構域的構象[27],增加其表達和活性,類固醇激素(如孕酮)對MR的親和力增加,導致腎臟鈉重吸收和鉀分泌,臨床表現為嚴重早發性高血壓、輕度低鉀血癥、血漿腎素活性降低和醛固酮低的妊娠加重的高血壓[14]。Geller綜合征的治療方法包括使用噻嗪類利尿劑、ENaC拮抗劑和輔以低鈉飲食。終止妊娠是使血壓正常化的有效治療方法[19]。
2.8 ?家族性糖皮質激素抵抗
家族性糖皮質激素抵抗也是一種罕見的綜合征,是由糖皮質激素受體上NR3C1基因的失活突變引起的。該突變導致糖皮質激素受體對皮質醇無反應[10,19],表現為糖皮質激素抵抗,即血漿ACTH和皮質醇增加、醛固酮水平低、低腎素、低鉀血癥、女性多毛癥、性早熟等。低劑量地塞米松治療可抑制下丘腦-垂體-腎上腺軸的活動,改善鹽皮質激素過多癥、皮質醇增多癥和高雄激素血癥。MR拮抗劑的使用,如螺內酯和依普利酮,有助于控制家族性糖皮質激素抵抗中的高血壓[10,19]。
2.9 ?高血壓伴短指(趾)畸形綜合征
高血壓伴短指(趾)畸形綜合征(hypertension and brachydactyly syndrome,HTNB)是一種罕見的常染色體顯性疾病,是由磷酸二酯酶3A(phosphodiesterase 3A,PDE3A)基因功能獲得突變引起的。該突變致使PDE3A酶活性增強,降解細胞內環磷酸腺苷或環磷酸鳥苷,加速血管平滑肌細胞增殖并導致高血壓[10]。臨床表現為身材矮小、腦血管異常、嚴重的非鹽依賴性高血壓。HTNB還可伴E型短指(趾)癥,其特征是掌骨和指骨增厚和縮短。HTNB也與延髓頭端中央外側的神經血管接觸有關,并可能導致壓力失調[27]。這種疾病在青春期前的兒童中很難診斷。如果不及時治療,患者50歲之前通常會發生卒中導致的死亡。使用β受體阻滯劑、α受體阻滯劑、鈣通道阻滯劑或血管緊張素轉化酶抑制劑/血管緊張素Ⅱ受體阻滯劑聯合或單藥治療可顯著降低高血壓,也可使用潛在具有PDE3A抑制作用的藥物[19,30]。
3 ?MH的診斷
目前MH尚無統一的診斷標準,主要依靠基因檢測的診斷(診斷流程可見 圖1)。為了提高基因檢測結果解讀的準確性,美國醫學遺傳學與基因組學會(The American College of Medical Genetics and Genomics,ACMG)編制了ACMG聯合共識作為序列變異解釋的標準和指南。

注:簡化MDRD公式計算,GFR=186×Scr﹣1.154×Age﹣0.203×0.742(女性)[GFR為腎小球濾過率,Scr為血清肌酐(mg/dL),Age為年齡(歲)][31-33]。
圖1??MH診斷流程圖
3.1??基因檢測的方法
隨著新一代測序技術的發展,越來越多的基因檢測方法比如Panel測序、全外顯子組測序(whole?exome sequencing,WES)、全基因組測序(whole?genome sequencing,WGS)等被應用于臨床。其中,WES可以捕獲所有的外顯子區域,包含了>85%的致病突變(包括非編碼區突變),適用于具有血緣關系或者具有嚴重癥狀,以及具有多個癥狀的患者,能顯著提高診斷率。但WES也存在特殊基因組區域(如:重復序列、假基因等)難以捕獲、特殊變異類型(復雜拷貝數變異、結構異常)難以識別等局限性[34]。而WGS可以對整個基因組檢測,檢測范圍全面,但是存在測序深度低、突變分析準確度低、對新變異結果的解讀困難等局限性[35]。因此需要臨床醫生選擇合適的測序方法,目前MH的診斷推薦采用診斷性基因組測序或WES,并在相應的家族成員中應用Sanger測序來確認診斷[19](見表2)。
3,2??ACMG指南的應用
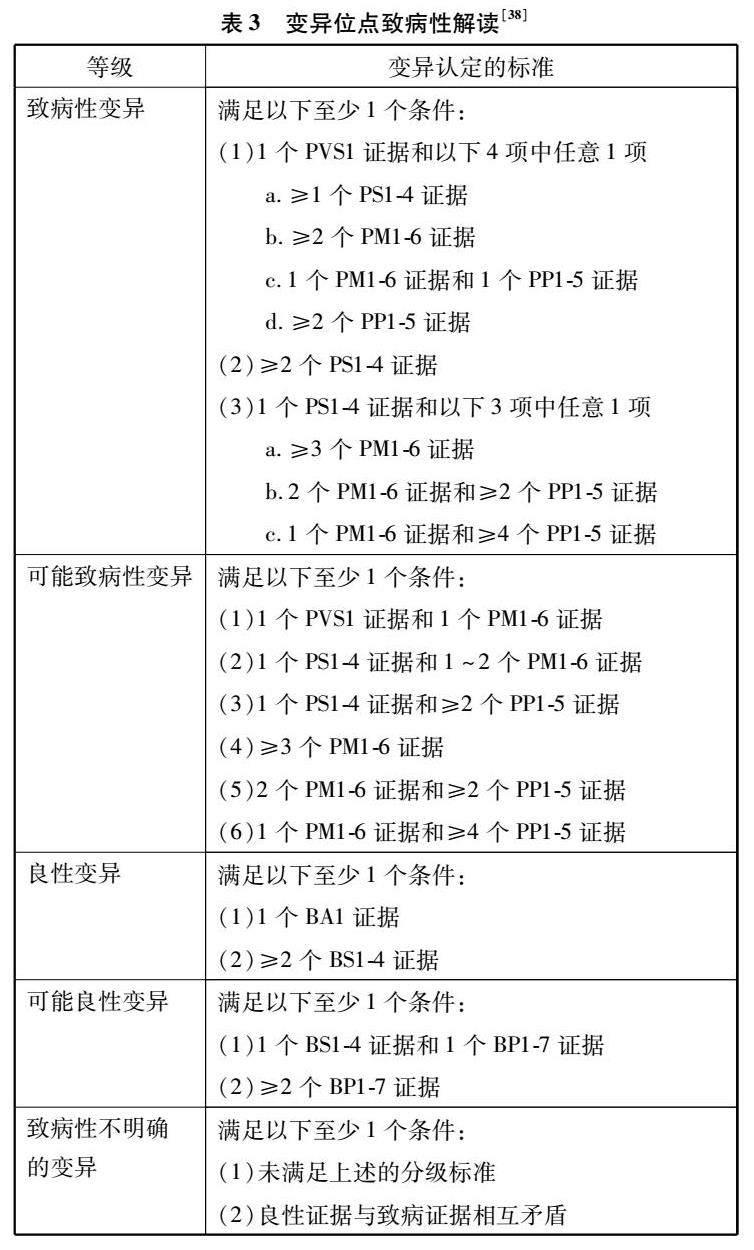
ACMG指南規范了MH診斷的流程和序列變體的解釋標準,包括標本的選取、基因變異的分級和變異分類。首先,臨床醫生根據預期用途選擇送檢樣本。若是產前可選擇羊水和絨毛細胞等;若是產后進行體質評估可選擇外周血,進行腫瘤性質評估可選用骨髓、外周血以及新鮮或冷凍的腫瘤組織等[36]。然后,檢測結果的解讀參照ACMG提供的兩套標準:一套用于致病性或可能致病性變異,對變異程度分為非常強(PVS1)、強(PS1-4)、中等(PM1-6)或輔助證據(PP1-5);另一套用于良性或可能良性變異,對變異程度分為獨立(BA1)、強(BS1-4)或輔助證據(BP1-6)。再根據組合分類標準將序列變體分為致病變異、可能致病變異、臨床未明變異、可能良性變異以及良性變異,其致病性呈逐級降低(見表3)。其中臨床未明變異又可分為臨床意義未明1~5級,這更需臨床醫生結合患者及其家族史、其他實驗室檢查結果等全面的臨床信息來評估變異。對于疑似隱性或新發變異,可將患者及其父母的樣本一起納入外顯子組測序中進行分析[37],這不僅可以增強風險評估,以最大限度地減少過度診斷,同時還可促進對真正疾病患者的適當診斷。最后,為患者制定個性化的治療方案,并定期跟蹤隨訪治療患者的血壓和生化指標,以監測長期療效并指導優化臨床方案。
4 ?MH的治療
MH患者暫無特異的治療,通常采用對癥治療(見表1)。由于MH遺傳模式的特異性以及發病機制的一致性,傳統的對癥降壓治療可能控制效果不佳。因此,在聯合使用傳統藥物的基礎上,開發針對特定基因位點的靶向藥物的精準治療成為高血壓治療的研究熱點,并且已經取得了顯著的成效。
反義寡核苷酸藥物IONIS-AGT-LRx,作用靶點是肝臟合成的血管緊張素原(angiotensinogen,AGT),可抑制AGT的合成,降低血管緊張素Ⅱ的水平。該藥物的Ⅱ期臨床試驗結果顯示,在基礎治療的背景上,能有效降低收縮壓和舒張壓[38]。小干擾RNA藥物zilebesiran,是一種化學修飾偶聯N-乙酰半乳糖胺的小干擾RNA,以AGT為靶點,抑制血管緊張素Ⅰ和Ⅱ的產生,從而達到降壓效果,通過每隔6個月皮下給藥的治療方案,也有可能提供比現有治療更明顯的優勢[39]。醛固酮合成酶抑制劑Baxdrostat,可抑制醛固酮的合成,從而降低血壓[40]。與此同時,血管緊張素1-7類似物、血管緊張素Ⅱ導向疫苗、奈普賴氨酸抑制劑、內皮素受體拮抗劑、多巴胺β-羥化酶抑制劑和腦氨肽酶A抑制劑等藥物正處于臨床開發的不同階段[41]。此外,經皮腎動脈交感神經消融術通過降低腎動脈交感神經活性,阻斷交感神經過度興奮,使血壓下降,也可以成為一種新的治療選擇[41]。動物實驗中使用CRISPR-Cas9基因編輯技術靶向介導肝臟AGT,可以持續降低血壓,提示此技術在將來可成為人類持續甚至終身控制高血壓的潛在療法[42]。
5 ?總結
MH的具體發病機制尚不能確定,臨床辨識困難且無統一的診治標準。現綜述了在此疾病中常見亞型的基因機制,強調了該病進行基因診斷的必要性,以及結合ACMG指南進行解讀分析的重要性。由于此病的復雜性和異質性,對相關患者群體進行遺傳學檢測和分析,將有助于更深層次地認識該疾病。
參?考?文?獻
Whelton PK,Flack JM,Jennings GLR,et al. Editors'?commentary on the 2023 ESH management of arterial hypertension guidelines[J]. Hypertension,2023,80(9):1795-1799.
Lu YT,Fan P,Zhang D,et al. Overview of monogenic forms of hypertension combined with hypokalemia[J]. Front Pediatr,2021,8:543309.
Raina R,Krishnappa V,Das A,et al. Overview of monogenic or Mendelian forms of hypertension[J]. Front Pediatr,2019,7:263.
Park SJ,Shin JI. Diagnosis and treatment of monogenic hypertension in children[J]. Yonsei Med J,2023,64(2):77-86.
Khandelwal P,Deinum J. Monogenic forms of low-renin hypertension:clinical and molecular insights[J]. Pediatr Nephrol,2022,37(7):1495-1509.
Travers S,Bouvattier C,Fagart J,et al. Interaction between accumulated 21-deoxysteroids and mineralocorticoid signaling in 21-hydroxylase deficiency[J]. Am J Physiol Endocrinol Metab,2020,318(2):E102-E110.
Wang W,Wei CJ,Cui XW,et al. Impacts of NF1 gene mutations and genetic modifiers in neurofibromatosis type 1[J]. 2021,12:704639.
de Blank PMK,Gross AM,Akshintala S,et al. MEK inhibitors for neurofibromatosis type 1 manifestations:clinical evidence and consensus[J]. Neuro Oncol,2022,24(11):1845-1856.
Nu?ez-Gonzalez L,Carrera N,Garcia-Gonzalez MA. Molecular basis,diagnostic challenges and therapeutic approaches of Bartter and Gitelman syndromes:a primer for clinicians[J]. Int J Mol Sci,2021,22(21):11414.
Ostrowska A,Skrzypczyk P. Monogenic hypertension[J]. Pol Merkur Lekarski,2022,50(297):198-201.
Semenova NA,Ryzhkova OR,Strokova TV,et al.Treti? slucha? sindroma pervichnogo al'dosteronizma, sudorog i nevrologicheskikh narusheni? (PASNA), obuslovlennogo variantom mutatsii de novo v gene CACNA1D[The third case report a patient with primary aldosteronism,seizures,and neurologic abnormalities (PASNA) syndrome de novo variant mutations in the CACNA1D gene][J]. Zh Nevrol Psikhiatr Im S S Korsakova,2018,118(12):49-52.
Araujo-Castro M,Martín Rojas-Marcos P,Parra Ramírez P. Familial forms and molecular profile of primary hyperaldosteronism[J]. Hipertens Riesgo Vasc,2022,39(4):167-173.
Funder JW,Carey RM,Mantero F,et al. The Management of Primary Aldosteronism:Case Detection,Diagnosis,and Treatment:An Endocrine Society Clinical Practice Guideline[J]. J Clin Endocrinol Metab,2016,101(5):1889-1916.
Scholl UI,St?lting G,Schewe J,et al.?CLCN2 chloride channel mutations in familial hyperaldosteronism type II[J]. Nat Genet,2018,50(3):349-354.
Mulatero P,Tauber P,Zennaro MC,et al. KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism[J]. Hypertension,2012,59(2):235-240.
Monticone S,Hattangady NG,Nishimoto K,et al. Effect of KCNJ5 mutations on gene expression in aldosterone-producing adenomas and adrenocortical cells[J]. J Clin Endocrinol Metab,2012,97(8):E1567-E1572.
Scholl UI,St?lting G,Nelson-Williams C,et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism[J].?Elife,2015,4:e06315.
Daniil G,Fernandes-Rosa FL,Chemin J,et al. CACNA1H mutations are associated with different forms of primary aldosteronism[J]. EBioMedicine,2016,13:225-236.
Lu Y,Fan P,Hakonarson H,et al. Monogenic hypertension-a type of "curable" hypertension[J]. Sci Bull (Beijing),2023,68(7):657-660.
Tetti M,Monticone S,Burrello J,et al. Liddle syndrome:review of the literature and description of a new case[J].?Int J Mol Sci,2018,19(3):812.
Kavinga Gunawardane PT,Grossman A. The clinical genetics of phaeochromocytoma and paraganglioma[J]. Arch Endocrinol Metab,2017,61(5):490-500.
Tsirlin A,Oo Y,Sharma R,et al. Pheochromocytoma:a review[J]. Maturitas,2014,77(3):229-38.
N?lting S,Ullrich M,Pietzsch J,et al. Current management of pheochromocytoma/paraganglioma:a guide for the practicing clinician in the era of precision medicine[J]. Cancers (Basel),2019,11(10):1505.
Sbardella E,Cranston T,Isidori AM,et al. Routine genetic screening with a multi-gene panel in patients with pheochromocytomas[J]. Endocrine,2018,59(1):175-182.
Patel D,Phay JE,Yen TWF,et al. Update on pheochromocytoma and paraganglioma from the SSO endocrine and head and neck disease site working group,part 2 of 2:perioperative management and outcomes of pheochromocytoma and paraganglioma[J]. Ann Surg Oncol,2020,27(5):1338-1347.
Lenders JWM,Eisenhofer G. Update on modern management of pheochromocytoma and paraganglioma[J]. Endocrinol Metab (Seoul),2017,32(2):152-161.
Levanovich PE,Diaczok A,Rossi NF. Clinical and molecular perspectives of monogenic hypertension[J]. Curr Hypertens Rev,2020,16(2):91-107.
Bulsari K,Falhammar H.Clinical perspectives in congenital adrenal hyperplasia due to 11β-hydroxylase deficiency[J]. Endocrine,2017,55(1):19-36.
Aggarwal A,Rodriguez-Buritica D. Monogenic hypertension in children:a review with emphasis on genetics[J]. Adv Chronic Kidney Dis,2017,24(6):372-379.
Toka O,Tank J,Sch?chterle C,et al. Clinical effects of phosphodiesterase 3A mutations in inherited hypertension with brachydactyly[J]. Hypertension,2015,66(4):800-808.
Levey AS,Bosch JP,Lewis JB,et al. A more accurate method to estimate glomerular filtration rate from serum creatinine:a new prediction equation. Modification of Diet in Renal Disease Study Group[J].?Ann Intern Med,1999,130(6):461-470.
Rule AD,Larson TS,Bergstralh EJ,et al. Using serum creatinine to estimate glomerular filtration rate:accuracy in good health and in chronic kidney disease[J]. Ann Intern Med,2004,141(12):929-937.
Levey AS,Coresh J,Greene T,et al.?Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate[J]. Ann Intern Med,2006,145(4):247-254.
Rexach J,Lee H,Martinez-Agosto JA,et al. Clinical application of next-generation sequencing to the practice of neurology[J]. Lancet Neurol,2019,18(5):492-503.
Wu S,Schmitz U. Single-cell and long-read sequencing to enhance modelling of splicing and cell-fate determination[J]. Comput Struct Biotechnol J,2023,21:2373-2380.
Shao L,Akkari Y,Cooley LD,et al. Chromosomal microarray analysis,including constitutional and neoplastic disease applications,2021 revision:a technical standard of the American College of Medical Genetics and Genomics (ACMG)[J]. Genet Med,2021,23(10):1818-1829.
Richards S,Aziz N,Bale S,et al. Standards and guidelines for the interpretation of sequence variants:a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med,2015,17(5):405-424.
Morgan ES,Tami Y,Hu K,et al. Antisense inhibition of angiotensinogen with IONIS-AGT-LRx:results of phase 1 and phase 2 studies[J]. JACC Basic Transl Sci,2021,6(6):485-496.
Desai AS, Webb DJ, Taubel J, et al. Zilebesiran, an RNA Interference Therapeutic Agent for Hypertension[J]. N Engl J Med. 2023,389(3):228-238.
Freeman MW,Halvorsen YD,Marshall W,et al.?Phase 2 trial of baxdrostat for treatment-resistant hypertension[J]. N Engl J Med,2023,388(5):395-405.
Ranasinghe P,Addison ML,Webb DJ. Small interfering RNA therapeutics in hypertension:a viewpoint on vasopressor and vasopressor-sparing strategies for counteracting blood pressure lowering by angiotensinogen-targeting small interfering RNA[J].?J Am Heart Assoc,2022,11(20): e027694.
Sun H,Hodgkinson CP,Pratt RE,et al. CRISPR/Cas9 mediated deletion of the angiotensinogen gene reduces hypertension:a potential for cure?[J].?Hypertension,2021,77(6):1990-2000.
收稿日期:2023-06-08
