多中心性網狀組織細胞增生癥1例并文獻復習
2024-05-09 12:01:24張秀君郭濤顧安康
中國醫藥指南 2024年12期
張秀君,郭濤,顧安康
1天津市中醫藥研究院附屬醫院皮膚科,天津 300120;2天津市中醫藥研究院附屬醫院病理科,天津 300120
網狀組織細胞增多癥(reticulohistiocytosis,RH)是一組罕見的單核吞噬細胞系統增殖性疾病,包括孤立性網狀組織細胞瘤、全身性網狀組織細胞增多癥和多中心網狀組織細胞增多癥(multicentric reticular histiocytosis,MRH)[1]。MRH 是RH 的一種常見亞型,它是一種炎癥性疾病,主要累及皮膚及骨關節,可引起皮膚變化和黏膜的病變,通常表現為丘疹樣或結節樣皮疹,并且可以模仿其他風濕性疾病,并引起破壞性關節炎。此外,MRH 也可累及內臟器官[2]。MRH沒有特異性的實驗室檢查結果,因此應根據其臨床表現、皮膚或滑膜活檢以及支持性影像學檢查結果進行診斷[3]。目前,MRH 沒有特效的治療方案,一般依靠經驗性治療,本病的預后與其骨關節損害及伴隨的疾病相關。本文報告1 例MRH,并進行文獻復習,旨在為今后的臨床診治提供指導。
1 病例資料
患者女性,43 歲,雙手甲皺多發暗紅色結節4 年。患者4 年前無明顯誘因雙手甲皺出現一芝麻粒大小暗紅色結節,無明顯自覺癥狀,未予處理,后皮疹逐漸增多,蔓延至前胸及耳郭。曾就診于當地醫院,予口服及外用藥治療(具體不詳),皮損未見好轉。患者既往雙手關節炎病史5 年,否認重工業及化學物質接觸史,否認家族類似疾病史。
體檢:一般情況好,系統檢查無異常。皮膚科檢查:雙手甲皺、前胸多發小結節,直徑0.3~0.5 cm,呈暗紅色,表面光滑,界限清,觸之質韌有彈性,雙手皮損呈對稱性分布,雙手關節活動輕微受限;耳郭孤立性淡紅結節,表面光滑,界限清,質韌(圖1)。
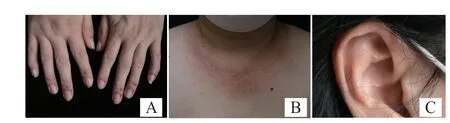
圖1 患者臨床皮損表現
實驗室及輔助檢查:血尿便常規、生化全項、免疫全項、類風濕因子三項等大致正常,心電圖及腹部B 超未見明顯異常;雙手X 線片:遠節指間關節面下骨質欠光整,關節面骨質硬化。
組織病理檢查:棘層輕度增厚,真皮內可見組織細胞數目增多,呈肉芽腫樣,并見多核巨細胞,胞質豐富,質均成細顆粒狀,呈“毛玻璃”樣(圖2),真皮淺層血管周圍可見較多的小淋巴細胞及中性粒細胞浸潤。PAS 染色組織細胞陽性。免疫組化:組織細胞CD68 陽性(圖3),S-100 和CD1a 陰性。診斷:多中心性網狀組織細胞增生癥。

圖2 組織病理表現
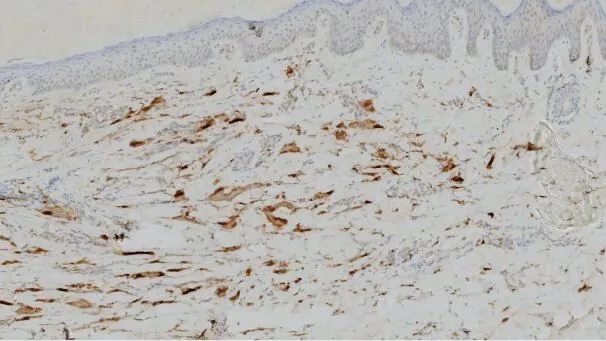
圖3 免疫組化染色CD68陽性(×100)
治療:予靜脈滴注甲強龍80 mg,每日1 次,治療5 天后口服強的松60 mg,每日1 次(逐漸減量),聯合口服甲氨蝶呤10 mg,1 次/周,硫酸羥氯喹0.2 g 每日1 次,治療后癥狀逐漸減輕;1 周后復查ESR6 mm/h、CRP3.9 mg/L;3 個月后結節、丘疹基本消退,未見關節疼痛。
2 討論
MRH 是網狀組織細胞增生癥的一種亞型,被歸類為Ⅱb 類非朗格漢斯細胞組織細胞增生癥,病變涉及皮膚和關節[4]。Weber 和Freudenthal 于1937 年首次報道本病[5]。本病為一種病因不明的多系統受累的全身性疾病,其特征是對稱性多關節炎和丘疹結節性皮膚病變,由于沒有明顯的生物學檢測標志,因此確診依賴于皮膚病理活檢及免疫組化染色。
2.1 臨床特點 本病好發于40~50 歲的成年女性,偶見兒童和老年人的報道[6]。關節癥狀是大部分患者就診時的首發癥狀,主要為進行性、對稱性、破壞性的多關節炎,常先累及雙手關節,表現為關節腫痛,可伴有晨僵和關節變形,可出現“風琴手”樣典型特征,經過一段時間的迅速惡化,關節炎通常可穩定,但不會明顯改善[7-9]。遠端指間關節最常受累,是MRH的典型特征之一,其他受累部位依次為膝、肩、腕、髖、踝、足、肘等處。本病患者100%有皮損,為組織細胞浸潤皮膚和黏膜所致,通常在關節受累后緩慢出現半透明棕紅色至肉色丘疹結節性病變,質地較硬,常見于手指及手腕關節,多見于手指背側,甲周病變具有典型的“珊瑚珠”外觀,累及面部可形成“獅子樣面容”,最終導致毀容。累及淋巴結、骨髓、肺、心內膜,累及心臟可能導致充血性心力衰竭及心包積液[7]。侵犯肌肉時常被誤診為皮肌炎。本文報道的患者皮疹后出現關節癥狀,遠節指間關節面下骨質欠光整,關節面骨質硬化,尚需隨診觀察。MRH 的病因尚不明確,可能與巨噬細胞活化及TNF-α、IL-12、IL-1和IL-6 水平升高有關[3]。MRH 與腫瘤及自身免疫性疾病有關[10-11]。約1/3 的MRH 患者伴有或繼發惡性腫瘤,其中70%以上患者本病的發生早于惡性腫瘤,因此MRH 被推測為一種副腫瘤性疾病,在診斷MRH 后應做癌癥篩查[12]。國內報道2 例分別合并胃癌及子宮內膜癌,l 例合并肺結核。合并血脂異常者亦較多見,國內報道共計5 例合并有血脂、血壓異常及心血管事件。MRH 和自身免疫性疾病可能表現出一定程度的臨床重疊,約1/3 的患者確診合并自身免疫性疾病,MRH相關的皮膚表現也可以模仿類風濕結節或皮肌炎相關的Gottron 丘疹。免疫功能障礙可能是MRH 的發病因素[13]。本例患者經系統檢查目前未發現合并腫瘤跡象。
2.2 實驗室及影像學檢查 實驗室指標對MRH 的診斷不具有特異性,30%~50%的患者會出現貧血、高脂血癥和急性期反應物的升高,有些患者有自身抗體陽性(如類風濕因子、抗環瓜氨酸抗體、抗核抗體、抗雙鏈DNA 抗體等)和細胞因子升高(如腫瘤壞死因子-α、白介素-6、單核細胞趨化蛋白-1)。影像學檢查主要表現為關節間隙增寬,X 線片可出現快速進展的糜爛性關節炎、軟骨缺失、彌漫性骨質疏松等[8]。
2.3 組織病理、免疫表型及超微結構 組織病理是MRH 的主要確診依據。典型的組織病理學檢可見真皮及皮下組織大量組織細胞及呈“毛玻璃”狀外觀的多核巨細胞,有時可見吞噬結締組織和細胞碎片現象。對PAS、蘇丹Ⅳ等呈陽性反應,耐淀粉酶[14]。
MRH 免疫組織化學通常顯示CD68(+),CD1a(-),S-100 蛋白(-),CD1a 與S-100 是朗格漢斯組織細胞標志物,本例陰性表明不是朗格漢斯細胞來源的組織細胞。
巨細胞邊緣有許多微絨毛,內含一個或數個高爾基體,富含線粒體、溶酶體、致密小體、吞噬體和髓磷脂;部分病例含有多形性胞漿內包涵體;巨細胞核不規則,常為分葉狀,電子密度中等,邊緣多個凹陷,有1~2 個核仁。
2.4 鑒別診斷 國內共有50 余例關于本病的個例報道,多數被誤診為類風濕關節炎和皮肌炎,另有個別曾按照環狀肉芽腫、麻風、結節病等治療,后經病理活檢及免疫組化染色確診為MRH,誤診率高達50%以上。MRH 需與網狀組織細胞瘤、皮肌炎及類風濕性關節炎相鑒別。①網狀組織細胞瘤:二者皮損形態相似,但網狀組織細胞瘤僅有皮膚及黏膜表現,不伴有發熱、破壞性多關節炎。②皮肌炎:MRH 可模仿皮肌炎相關的Gottron 丘疹、披肩征等皮炎樣特征,并可伴有光敏性,但無進行性對稱性近端肌無力等肌炎相關表現。此外,可通過肌炎特異性抗體、免疫全項、組織病理等與無肌病性皮肌炎相鑒別。③類風濕性關節炎:類風濕性關節炎主要表現為手、足小關節的多關節、對稱性、侵襲性關節炎癥,部分可出現皮下結節,長期可導致關節變形,MRH 與其臨床表現相似,但類風濕性關節炎一般伴有類風濕因子異常,借此可相鑒別。盡早進行皮損部位的病理活檢及免疫組化染色是減少誤診的重要因素。
2.5 治療及預后 本病的預后與骨關節表現、患者自身免疫性疾病和腫瘤有關。MRH 早期準確診斷是預防糜爛性關節炎進展為殘缺性關節炎的基礎,定期進行免疫性疾病及腫瘤疾病的篩查也是必要的。目前還沒有針對MRH 的標準化治療方法,主要依靠經驗性治療。常用治療方式為口服糖皮質激素、非甾體抗炎藥或免疫抑制劑。在疾病控制不佳、使用糖皮質激素或合并骨質減少的情況下,可考慮將雙膦酸鹽類藥物作為附加藥物以控制骨關節炎的發展[6]。Zhao 等[15]報道,依那西普、英夫利昔單抗和阿達木單抗等TNF抑制劑也是本病的替代治療藥物。近來年,JAK 抑制劑,如托法替尼和烏帕替尼也被用于本病的治療,可明顯改善皮膚及關節病變[4]。
綜上所述,MRH 在臨床上較為罕見,其發病原因不明,除皮膚及關節損害外,還可累及其他組織或內臟。本病的確診需要多種方法進行綜合判斷,病理學檢查是金標準,早期和準確診斷對預防日常生活活動受損至關重要。本病還可合并多種惡性腫瘤,因此應早期進行腫瘤篩查;MRH 與結締組織病的表現類似,又可以與其同時出現,因此要注意鑒別診斷,避免誤診、漏診。激素聯合免疫抑制劑治療是MRH 最常見的治療方案,可以改善患者的大部分臨床癥狀,當常規藥物效果不理想時可考慮生物制劑。皮膚病變可以通過二氧化碳激光應用或手術切除來治療。
