超高效液相色譜串聯質譜法測定海參中硝基呋喃類代謝物殘留量
2024-10-17 00:00:00寧晨
食品安全導刊·中旬刊 2024年9期
摘 要:目的:建立一種快速、準確、高效的超高效液相色譜串聯質譜檢測海參中硝基呋喃類代謝物殘留量的方法。方法:采用40 ℃水浴中超聲1 h,45 ℃氣浴中衍生5 h,pH值7.0~7.5條件進行樣品前處理,以2 mmol·L-1乙酸銨的0.1%甲酸水溶液-乙腈為流動相進行梯度洗脫,超高效液相色譜串聯質譜測定。結果:4種硝基呋喃類代謝物在0.5~20.0 ng·mL-1線性關系良好,加標平均回收率在97.3%~103.6%,方法檢出限為0.5 μg·kg-1,定量限為1.0 μg·kg-1。結論:該方法可用于市售海參中硝基呋喃類代謝物殘留量的快速檢測。
關鍵詞:海參;硝基呋喃類代謝物;超高效液相色譜串聯質譜法
Determination of Nitrofuran Metabolites in Sea Cucumber by Ultra-high Performance Liquid Chromatography Tandem Mass Spectrometry
NING Chen
(Ningde Quality Inspection Institute of Product, Ningde 352100, China)
Abstract: Objective: To establish a rapid, accurate, and efficient method for detecting residual nitrofuran metabolites in sea cucumber using ultra-high performance liquid chromatography tandem mass spectrometry. Method: Sample pretreatment was carried out using ultrasound for 1 hour in a 40 ℃ water bath, derivatization for 5 hours in a 45 ℃ gas bath, and pH values ranging from 7.0 to 7.5. Gradient elution was performed using a mobile phase of 2 mmol·L-1ammonium acetate in 0.1% formic acid aqueous solution acetonitrile, and ultra-high performance liquid chromatography tandem mass spectrometry was used for determination. Result: The four nitrofuran metabolites showed a good linear relationship in the range of 0.5 ng·mL-1 to 20.0 ng·mL-1, with an average recovery rate of 97.3% to 103.6%. The detection limit of the method was 0.5 μg·kg-1, and the quantification limit was 1.0 μg·kg-1. Conclusion: This method can be used for rapid detection of residual nitrofuran metabolites in commercially available sea cucumbers.
Keywords: sea cucumber; nitrofuran metabolites; ultra high performance liquid chromatography-tandem mass spectrometry
海參作為一種富含多種人體必需營養成分的海洋產品[1-2],隨著人們生活水平的提高,已逐漸成為普通家庭餐桌上的常見美食。然而,在海參養殖過程中,為防止出現病害,常使用全池潑灑法進行水體消毒[3]。硝基呋喃類藥物是常用的一種殺菌劑,其具有抗菌譜廣、效果明顯且價格便宜等優勢,被海參養殖戶廣泛用于預防在養殖過程中出現的病害[4-5]。
硝基呋喃藥物的殘留物可能通過食物鏈進入人體,對健康構成潛在風險[6-7]。目前,中華人民共和國農業農村部公告第250號(食品動物中禁止使用的藥品及其他化合物清單)規定,硝基呋喃類藥物屬于禁用獸藥,在海參等水產品中不得檢出。因此,加強對硝基呋喃類代謝物等獸藥殘留的檢測,是保障水產品質量安全的重要措施。
現行國家標準在檢測海參中的硝基呋喃類代謝物時,通常采用液相色譜-串聯質譜法,包括進行酸解、衍生、凈化等前處理步驟,檢測周期較長,通常需要1~2 d[8-10]。而快檢方法又存在假陽性的風險,難以滿足海參交易中的檢測需求[11-12]。因此,建立一種可快速出具結果的檢測方法,顯得至關重要。本文旨在研究一種快速、準確、高效檢測海參中硝基呋喃類代謝物的方法,為水產品質量安全與行業發展提供技術支撐。
1 材料與方法
1.1 材料與試劑
乙酸乙酯、乙腈、甲醇和乙酸銨,均為色譜純;2-硝基苯甲醛為優級純;濃鹽酸、無水磷酸氫二鉀,均為分析純。
標準品:3-氨基-2-惡唑烷基酮(AOZ,純度99.14%,CAS NO.80-65-9)、5-嗎啉甲基-3-氨基-2-惡唑烷基酮(AMOZ,純度99.19%,CAS NO.43056-63-9)、1-氨基-2-內酰脲鹽酸鹽(AHD·HCl,純度99.90%,CAS NO.2827-56-7)和氨基脲鹽酸鹽(SEM·HCl,純度99.77%,CAS NO.563-41-7),均購自Dr.Ehrenstorfer公司。
內標:AOZ-D4(純度99.7%,CAS NO.1188331-23-8)、AMOZ-D5(純度98.0%,CAS NO.1017793-94-0)、AHD-13C3(純度98%,CAS NO.957509-31-8)和SEM·HCl-13C,15N2(不確定度±3%,k=2,CAS NO.1173020-16-0),均購自BePure公司。
以日常檢測積累的海參陽性樣品作為測試樣,選取了3個陽性樣品(樣品1、樣品2、樣品3)。其中,樣品1為含AOZ和AHD的海參陽性樣品,AOZ和AHD的含量分別為10.2 μg·kg-1和4.1 μg·kg-1;樣品2為含SEM的海參陽性樣品,SEM的含量為19.9 μg·kg-1;樣品3為含AMOZ的海參陽性樣品,AMOZ的含量為3.5 μg·kg-1。
1.2 儀器與設備
Agilent LC1290-G6460A液相色譜-串聯質譜儀(配電噴霧離子源);VORTEX-5渦旋振蕩器,海門其林貝爾;JZQ-Ⅱ均質器,天津津德;TGL-15B高速離心機,上海安亭;ZD-85氣浴恒溫振蕩器,常州國華電器;SE812氮吹儀,北京帥恩;KQ-500DE超聲波清洗器,昆山超聲儀器。
1.3 實驗方法
1.3.1 標準溶液配制
混合標準儲備液(1.0 mg·mL-1):精確稱量AOZ、AMOZ、AHD和SEM標準品各0.010 0 g于10 mL容量瓶中,加1 mL純水溶解,用乙腈定容。
混合內標儲備液(10.0 μg·mL-1):精確移取AOZ-D4、AMOZ-D5、AHD-13C3和SEM·HCl-13C,15N2內標物各100 μL,加900 μL乙腈。
標準工作溶液:空白海參樣品按照樣品前處理步驟進行衍生、萃取得下清液,準確移取適量4種硝基呋喃類代謝物混合標準中間溶液(100 ng·mL-1),用下清液稀釋得到濃度為0.5 ng·mL-1、1.0 ng·mL-1、2.0 ng·mL-1、5.0 ng·mL-1、10.0 ng·mL-1和20.0 ng·mL-1的標準工作溶液。同時,準確移取適量的100.0 ng·mL-1硝基呋喃內標工作溶液,使得標樣中最終內標濃度為5.0 ng·mL-1。
1.3.2 樣品前處理
稱取已均質的海參樣品2.0 g(精確至0.001 g)于50 mL離心管中,準確加入100 μL混合內標工作液(100.0 ng·mL-1),加入0.5 mol·L-1的鹽酸溶液5.0 mL和0.1 mol·L-1的2-硝基苯甲醛溶液200 μL,渦旋混合10 min,于超聲波清洗器中40 ℃水浴超聲1 h,然后置于氣浴恒溫振蕩器中,45 ℃避光衍生5 h。
取出已衍生的樣品,冷卻至室溫,緩慢加入1.0 mol·L-1的K2HPO4溶液,調節溶液的pH值為7.0~7.5,加入乙酸乙酯20.0 mL萃取,低速渦旋混合30 s,平行振蕩10 min后,于高速離心機中10 000 r·min-1離心5 min。取上清液至氮吹管中,放入氮吹儀40 ℃水浴中氮氣吹干(在氮吹過程中,應留意提取液是否被吹干,若已干應及時取出氮吹管,否則會影響測定結果)。移取2.0 mL乙腈-0.1%甲酸水溶液(1+9,V+V)加入已干的氮吹管中,再加入2.0 mL乙腈飽和的正己烷,低速渦旋溶解殘留物,以10 000 r·min-1離心10 min,下清液過有機系尼龍過濾器(0.22 μm,13 mm)至進樣瓶,供液相色譜-串聯質譜儀檢測。
1.3.3 液相色譜和質譜條件
(1)色譜條件。色譜柱:ACQUITY UPLC BEH C18(100 mm×2.1 mm,1.7 μm);柱溫:30 ℃;進樣量:10 μL;流速:0.3 mL·min-1;流動相A:含2 mmol·L-1乙酸銨的0.1%甲酸水溶液,B:乙腈。洗脫條件:0~0.6 min,10%→20%B;0.6~3.0 min,20%B;3.0~6.0 min,20%→25%B;6.0~7.0 min,25%→60%B;7.0~8.0 min,60%→95%B;8.0~10.0 min,95%B;10.0~10.1 min,95%→10%B;10.1~12.0 min,10%B。
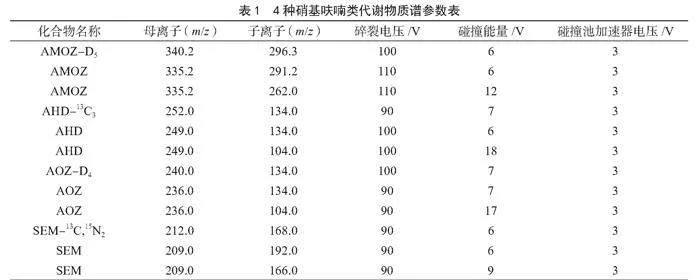
(2)質譜條件。電噴霧離子源類型:ESI+;干燥氣溫度:325 ℃;干燥氣流量:10 L·min-1;鞘氣溫度:350 ℃;鞘氣流量:12 L·min-1;霧化器壓力:45 psi;毛細管電壓:4 500 V。4種硝基呋喃類代謝物質譜參數見表1。
2 結果與討論
2.1 前處理條件優化
2.1.1 超聲時間優化
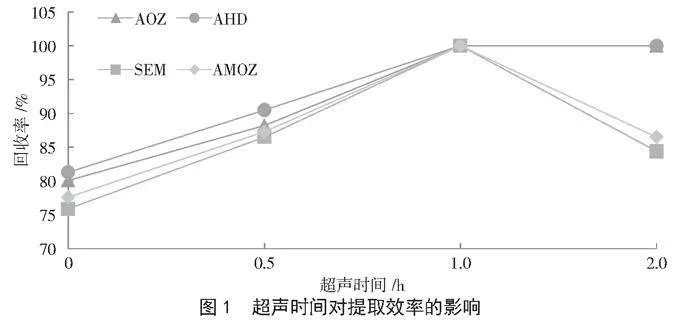
超聲提取是一種在較低溫度下進行的高效、適用性廣的前處理技術[13-14]。為進一步優化超聲時間,本研究固定1.3.2的樣品前處理條件,探究超聲時間0 h、0.5 h、1.0 h和2.0 h對4種硝基呋喃類代謝物回收率的影響。由圖1可知,超聲時間1 h時,4種硝基呋喃類代謝物的回收率達到100%,而當超聲時間超過1 h時,AOZ、AHD提取效率不受影響,SEM、AMOZ提取效率呈下降趨勢,因此,本研究選擇最佳超聲時間為1 h。
2.1.2 衍生時間優化
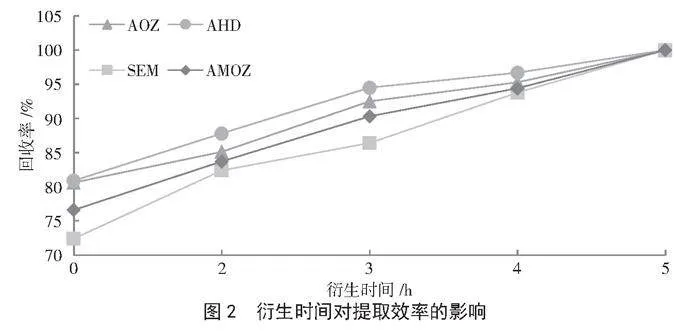
固定1.3.2的樣品前處理條件,探究衍生時間0 h、2 h、3 h、4 h和5 h對4種硝基呋喃類代謝物回收率的影響。由圖2可知,隨著衍生時間的延長,4種硝基呋喃類代謝物的回收率均呈上升趨勢,當衍生時間為5 h時,AOZ、AHD、SEM和AMOZ的回收率均達到100%。因此,本研究選擇最佳衍生時間為5 h。
2.1.3 衍生溫度優化
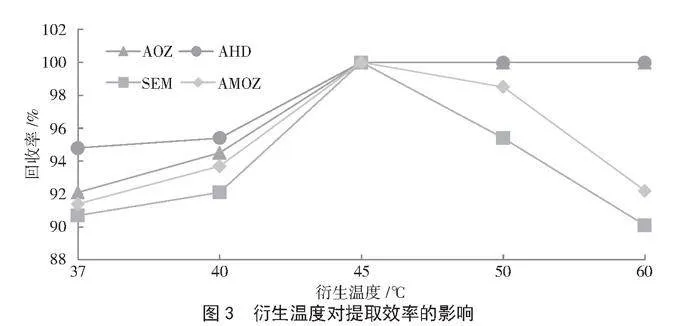
固定1.3.2的樣品前處理條件,探究衍生溫度37 ℃、40 ℃、45 ℃、50 ℃和60 ℃對4種硝基呋喃類代謝物回收率的影響。由圖3可知,衍生溫度在45 ℃時,AOZ、AHD、SEM和AMOZ的回收率均達到100%,但當衍生溫度繼續升高時,SEM、AMOZ的回收率呈現下降趨勢,這可能是由于溫度升高會引起SEM、AMOZ這2種代謝物的降解。因此,本研究選擇最佳衍生溫度為45 ℃。
2.2 色譜條件優化
本研究采用ACQUITY UPLC BEH C18色譜柱,探究了0.1%甲酸水溶液-乙腈、0.1%甲酸水溶液-甲醇、2 mmol·L-1乙酸銨水溶液-乙腈、2 mmol·L-1乙酸銨水溶液-甲醇、含2 mmol·L-1乙酸銨的0.1%甲酸水溶液-乙腈和含2 mmol·L-1乙酸銨的0.1%甲酸水溶液-甲醇這6種流動相的分離及出峰效果。結果表明,含乙酸胺的流動相使得4種硝基呋喃類代謝物及內標的色譜峰型更加的穩定,特別是AOZ不會再出現裂峰;而含甲酸流動相的目標物分離效果更好,出峰時間間隔較長;甲醇體系的流動相雖然色譜峰更尖銳,但會導致質譜系統的壓力比使用乙腈時高出約100 bar,對設備和色譜柱的損耗較大。因此,本研究選擇含2 mmol·L-1乙酸銨的0.1%甲酸水溶液-乙腈作為流動相。
2.3 方法驗證
2.3.1 線性方程和檢出限
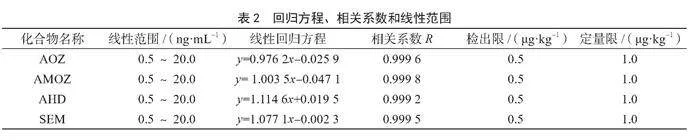
取通過1.3.1方法得到的系列標準工作溶液,按照1.3.3液質條件進行測定,以峰面積與內標峰面積的比值為縱坐標,經過衍生的樣品質量濃度與內標濃度的比值為橫坐標,繪制標準曲線,并計算回歸方程和相關系數R,以3倍信噪比計算方法檢出限,10倍信噪比計算方法定量限。由表2可知,4種硝基呋喃類代謝物在0.5~20.0 ng·mL-1線性關系良好,相關系數R均大于0.999,檢出限為0.5 μg·kg-1,定量限為1.0 μg·kg-1。
2.3.2 回收率和精密度
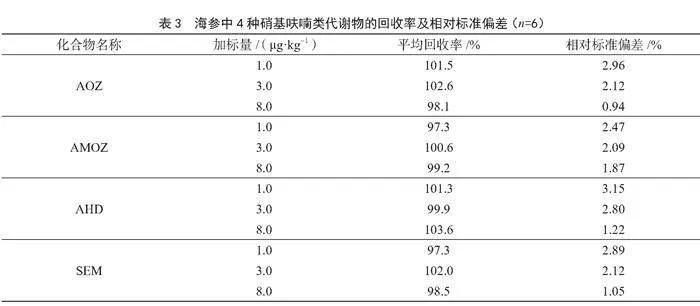
稱取海參陰性樣品2.00 g(精確至±0.01 g),加標水平濃度分別為1.0 μg·kg-1、3.0 μg·kg-1、8.0 μg·kg-1,每個水平濃度平行測定6次,按1.3.2和1.3.3進行樣品前處理和測定。依據內標法計算回收率和相對標準偏差。由表3可知,AOZ、AMOZ、AHD和SEM的平均回收率在97.3%~103.6%,相對標準偏差在0.94%~3.15%,表明該方法回收率和精密度良好。
2.4 實際樣品測定
采用本文建立的方法檢測在養殖海域采集的100批次海參樣品,結果表明,海參樣品共檢測出呋喃唑酮類代謝物11批次,其中,有4批次檢測出AHD,2批次檢測出SEM。說明目前海參中仍存在硝基呋喃類藥物的污染,養殖戶仍有違規使用禁用藥物硝基呋喃的情況,這一情況需要引起相關部門的高度重視,并采取相應措施。
3 結論
本文通過對樣品前處理中超聲時間、衍生時間、衍生溫度,以及儀器條件中流動相的優化,建立了一種超高效液相色譜串聯質譜快速、準確測定海參中硝基呋喃類代謝物的檢測方法,通過方法學驗證評估,該方法具有準確度高、回收率和精密度良好的優勢,同時可將檢測時間縮短至7~9 h,且操作簡便,無須昂貴的前處理設備,滿足海參交易市場的檢測需求,有助于保障海參產品的質量安全和促進水產品行業的可持續發展。
參考文獻
[1]韋豪華,張紅玲,李興太.海參化學成分及生物活性研究進展[J].食品安全質量檢測學報,2017,8(6):2054-2061.
[2]王婧媛,王聯珠,孫曉杰,等.海參加工工藝、營養成分及活性物質研究進展[J].食品安全質量檢測學報,2018,9(11):2749-2755.
[3]胡軍娜,馬娟,余煒.水產養殖病害特點及防治技術[J].河南水產,2019(4):13-15.
[4]戴欣,李改娟.水產品中硝基呋喃類藥物殘留的危害、影響以及控制措施[J].吉林水利,2011(9):61-62.
[5]郭添榮,韓世鶴,吳文林,等.UPLC-MS/MS法快速檢測人工養殖淡水魚中硝基呋喃類代謝物[J].環境化學,2022,41(11):3534-3543.
[6]王群,呂海燕,宋懌.硝基呋喃類在水產品及其養殖環境中的消解規律研究進展[J].中國漁業質量與標準,2014,4(6):16-21.
[7]胡家勇,柳鑫,張莉,等.食品中硝基呋喃類藥物殘留檢測技術研究進展[J].食品安全質量檢測學報,2024,15(4):148-159.
[8]邢麗紅,李兆新,孫偉紅,等.液相色譜-串聯質譜法檢測水產品中硝基呋喃類藥物的殘留量[J].食品安全質量檢測學報,2017,8(4):1233-1239.
[9]張秀妍,馬琳,王慧龍.海參和海參苗種中硝基呋喃類代謝物殘留的液質聯用檢測法[J].口岸衛生控制,2012,17(6):24-28.
[10]楊鵬.水產品中硝基呋喃類藥物及其代謝物的LC-MS/MS檢測方法研究和應用[D].衡陽:南華大學,2019.
[11]于丁一,嚴忠雍,朱敬萍,等.四合一膠體金試紙條快速檢測水產品中的硝基呋喃代謝物[J].理化檢驗(化學分冊),2019,55(3):364-367.
[12]胡金春,張曉輝.水產品中氯霉素及四種硝基呋喃代謝物殘留快速檢測方法研究[J].福建分析測試,2016,25(6):1-4.
[13]王猛,王康康,楊雪麗,等.超聲輔助衍生—液相色譜串聯質譜法快速檢測魚肉中4種硝基呋喃類代謝物殘留[J].疾病預防控制通報,2020,35(3):28-31.
[14]李招,李劍,徐巧.UPLC-MS/MS法測定雞肉中5種硝基呋喃代謝物[J].食品工業,2023,44(9):282-286.
