
對藏藥四味雪蓮花顆粒中含量測定方法的改進
2010-02-07 03:48:18張秀峰
中成藥 2010年12期
張秀峰
(青海省藥品檢驗所,青海西寧810000)
四味雪蓮花顆粒收載于《國家中成藥標準匯編》(中成藥地方標準上升國家標準部分)內科心系合成分冊[1]。由紅景天、雪蓮花、大黃、蕨麻四味藏族傳統用藥組成。藏醫用于三大因素平衡紊亂,隆,培根功能失調,氣血上升,血瘀痰阻所致的高脂血癥[1]。原標準中含量測定為高效液相色譜法測定大黃素含量,本實驗用高效液相色譜法同時測定大黃素、大黃酚的總量。此法簡便、穩定、重現性好,能夠更好的控制四味雪蓮花顆粒的質量。
1 儀器與試藥
Agilent1100高效液相色譜儀,Agilent G1314 VWD紫外檢測器,HP化學工作站(美國Agilent公司);電子天平(SartoriusBT224S,北京賽多力斯儀器公司);電子天平(SartoriusR200-D,北京賽多力斯儀器公司);電子天平(METTLER PB1502-S,瑞士 METTLER公司);超聲波清洗器(B2500s-MT,上海必能信超聲波清洗儀有限公司)。
大黃素對照品(110756-200110),大黃酚對照品(110796-200412)均由中國藥品生物制品檢定所提供,為含量測定用。甲醇為色譜純,其余試劑均為分析純,水為重蒸餾水;四味雪蓮花顆粒樣品與陰性藥材由青海金訶藏藥藥業有限責任公司提供,樣品批號分別為:081101、081102、081103。
2 方法與結果
2.1 色譜條件與系統適用性試驗 色譜柱:依利特Hypersil-ODS2 柱(250 mm ×4.6 mm,粒徑 5 μm);柱溫:室溫;流動相:甲醇-0.1%磷酸溶液(80∶20);檢測波長:254 nm;理論板數按大黃素峰計算,大于10 000(應不低于3 000)。
在上述色譜條件下,得到四味雪蓮花顆粒供試品及大黃素、大黃酚對照品色譜圖,大黃素的保留時間在8.5 min左右,大黃酚的保留時間在11.3 min左右。
2.2 對照品溶液的制備 精密稱取大黃素對照品9.93 mg,大黃酚對照品27.55 mg分別置250 mL量瓶中,加甲醇溶解并稀釋至刻度,搖勻;分別精密量取上述大黃素對照品溶液4 mL,大黃酚對照品溶液2 mL置100 mL量瓶中,加甲醇稀釋置刻度,搖勻即得混合對照品溶液(大黃素對照品溶液濃度為1.588 μg/mL,大黃酚對照品溶液濃度為2.204 μg/mL)。
2.3 供試品溶液制備 取本品裝量差異下內容物約0.5 g,精密稱定,置具塞錐形瓶中,精密加入甲醇25 mL,稱定重量,加熱回流45 min,放冷,再稱定重量,用甲醇補足減失的重量,搖勻,濾過。精密量取續濾液10 mL,置100 mL錐形瓶中,揮去溶劑,加8%鹽酸液10 mL,超聲處理2 min(50 Hz,120 W),再加三氯甲烷10 mL,加熱回流 1 h,放冷,移至分液漏斗中,用少量三氯甲烷洗滌容器,并入分液漏斗中,分取三氯甲烷層,酸液再用三氯甲烷提取3次,每次約10 mL,合并三氯甲烷液,減壓回收溶劑至干,殘渣加甲醇使溶解,轉移至10 mL量瓶中,加甲醇至刻度,搖勻,濾過,取續濾液作為供試品溶液。
2.4 陰性對照溶液的制備 按照處方的組成,取除大黃外的其余藥味,按制備工藝要求制成不含大黃的制劑,按供試品溶液制備項下的方法制備陰性對照溶液。在上述色譜條件下,精密吸取供試品溶液、對照品溶液、陰性對照溶液各10 μL,分別注入液相色譜儀。供試品色譜圖中,在與大黃素、大黃酚對照品色譜圖相應保留時間上,分別有相同的色譜峰,而陰性對照在此無干擾。
2.5 線性考察 分別精密吸取上述已配制好的大黃素(濃度為1.588 μg/mL)與大黃酚(濃度為2.204 μg/mL)對照品混合溶液 2、4、8、12、16、20 μL,按上述條件分別注入色譜儀測定,以對照品溶液進樣量(μg)為橫坐標,以峰面積積分值為縱坐標,繪制標準曲線,計算回歸方程得:大黃素Y=6.081 1X-0.104 7,r=0.999 9,大黃素進樣量在 0.003 176~0.031 76 μg范圍內與峰面積呈線性關系;大黃酚Y=11.883X -0.957 8,r=0.999 9,大黃酚進樣量在0.004 41~0.044 1 μg范圍內與峰面積呈線性關系。
2.6 精密度考察 精密吸取上述已配制好的大黃素對照溶液(濃度為1.588 μg/mL),大黃酚對照溶液(濃度為2.204 μg/mL),連續進樣5次,每次進樣量為10 μL,按色譜條件操作進行,測峰面積,以峰面積計算大黃素RSD=0.3%,大黃酚RSD=0.9%,結果表明:大黃素、大黃酚測定方法精密度良好。
2.7 穩定性試驗 取供試品溶液(批號:081103),按上述色譜條件分別于 0、2、4、6、8、12 h 進樣量 10 μL,測定峰面積的大黃素RSD=3.0%,大黃酚RSD=1.3%,說明供試品溶液在12 h內穩定。
2.8 重復性試驗 按照上述含量測定方法對相同批號(081103)樣品制備5份供試液,分別進樣10 μL,測定峰面積,結果大黃素RSD=1.6%,大黃酚RSD=1.4%,說明本方法重現性良好。
2.9 加樣回收率試驗 精密稱取大黃素2.55 mg,大黃酚4.28 mg置250 mL量瓶中,加甲醇使溶解并稀釋至刻度,配制成每1 mL含大黃素為0.010 2 mg,大黃酚為0.017 10 mg的混合溶液。取已知含量的四味雪蓮花顆粒樣品(批號081103,大黃素含量為0.064 2 mg/g,大黃酚含量為0.102 9 mg/g)0.25 g,稱取9份,置具塞錐形瓶中,分別精密加入混合對照品溶液1、1.5、2.5 mL各3份,按上述供試品溶液的制備方法制備,按上述色譜條件測定大黃素與大黃酚的含量,計算回收率。結果平均回收率大黃素為98.1%,RSD=2.1%;大黃酚為98.9%,RSD=2.3%。結果表明本方法加樣回收較好,結果見表1、2。
2.10 樣品測定 按上述含量測定方法,對3批樣品中所含大黃素、大黃酚的總量進行含量測定,結果見表3。
2.11 原方法測定結果與改進后方法測定結果比較 原標準測定結果與改進后的方法測定結果見表4。
測定結果表明,原標準中大黃素含量低于萬分之一,改進后測定結果及數據均高于萬分之一,對于制劑質量的控制更有意義。建議將原標準中單測定大黃素含量改進為同時測定大黃素、大黃酚的含量。

表1 大黃素回收率測定結果
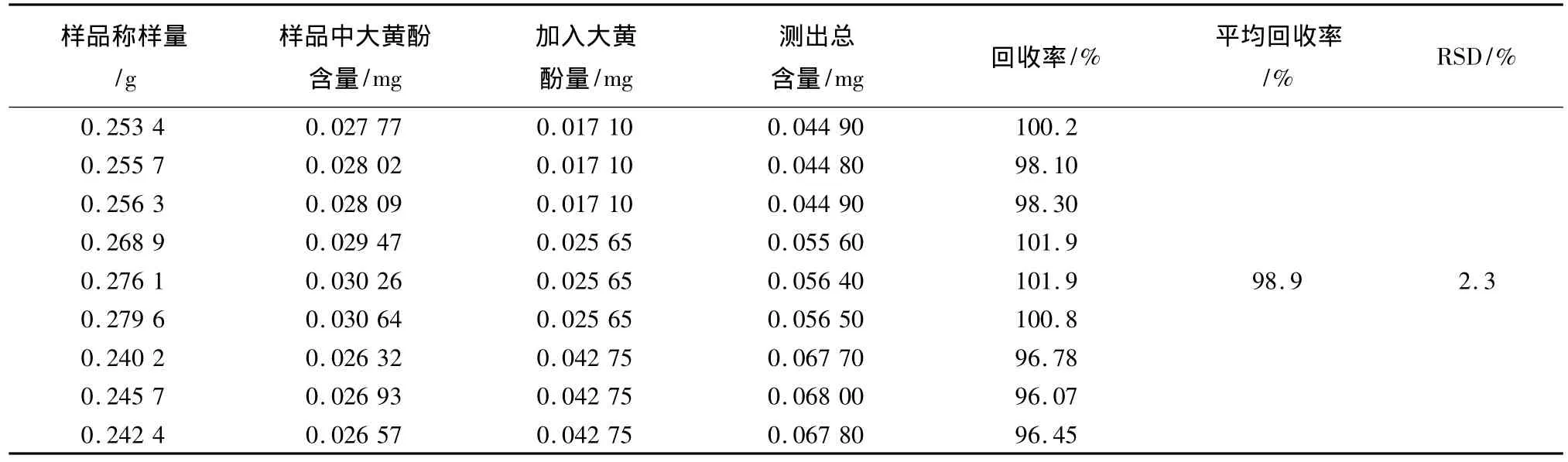
表2 大黃酚回收率測定結果
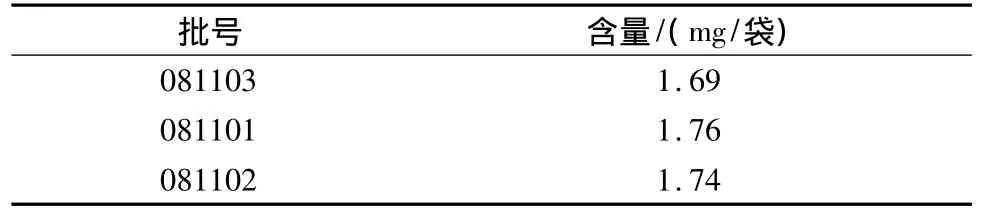
表3 樣品測定結果
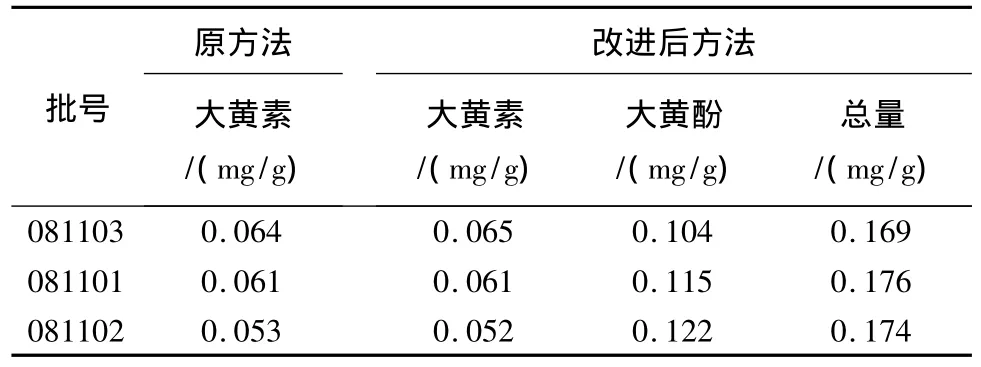
表4 2種測定方法結果比較
3 討論
3.1 本次試驗對大黃素和大黃酚進行紫外全波長掃描,大黃素、大黃酚在254 nm和436 nm處都有最大吸收,參照2005版《中國藥典》一部[2]大黃藥材的含量測定項,選擇波長為254 nm進行檢測,樣品主峰分離度較好,且陰性對照無干擾。
3.2 參照2005版《中國藥典》[2]一部中大黃藥材含量測定方法測定四味雪蓮花顆粒樣品,色譜峰分離度和峰形并不理想,后改用甲醇-0.1%磷酸溶液(80∶20)作為流動相,20 min內出峰完成,且分離度較好。
3.3 在試驗過程中,我們考察了超聲30、45、60、75 min和回流30、45、60、75 min提取樣品中大黃素和大黃酚的含量,發現回流的樣品含量總體高于超聲提取,且45 min和60 min,75 min大黃素、大黃酚的含量相差不大,所以本研究回流45 min作為本次樣品的提取方式。
[1]國家中成藥標準匯編(中成藥地方標準上升國家標準部分)內科心系分冊[S].2002:156-159.
[2]中國藥典[S].一部.2005:17-18.
