微生物法制備單唾液酸四己糖神經節苷脂的機制和方法
2010-07-13 09:05:16董惠鈞姜俊云
中國醫藥生物技術 2010年2期
董惠鈞,姜俊云
?
微生物法制備單唾液酸四己糖神經節苷脂的機制和方法
董惠鈞,姜俊云
273400 山東省臨沂,魯南制藥集團股份有限公司
單唾液酸四己糖神經節苷脂(GM1)是含有單個唾液酸的酸性鞘糖脂,被認為是除腦源性神經生長因子和層粘蛋白外,對神經系統損傷具有顯著臨床療效的生物藥物。GM1主要來源于哺乳動物含豐富神經的腦組織,經有機溶劑提取和柱層析分離精制獲得。由于 GM1在腦組織神經節苷脂類物質中的含量非常少約為 32.9%[1],提取收率低,所以要經過多唾液酸神經節苷脂的轉化作用生成單唾液酸四己糖神經節苷脂。目前轉化方法主要是酸法轉化,但是酸法轉化的效率低,處理溫度高以及處理量少,限制了其大規模應用。近年來,科研人員開發了一種利用酶促反應水解唾液酸糖苷鍵進行神經節苷脂轉化的方法,該法具有轉化效率高、轉化條件溫和、處理量大等優點。本文概括介紹了國內外在神經節苷脂、唾液酸酶和 GM1微生物酶法轉化工藝研究等方面的研究進展,并展望了生物法制備 GM1的發展趨勢。
1 神經節苷脂結構性質與功能
1.1 結構性質
神經節苷脂(ganglidosides,GLS)由一類含唾液酸的親水寡糖通過 β-1, 1 糖苷鍵與疏水的神經酰胺連接而成的兩性大分子(圖1)。神經節苷脂由鞘氨醇、脂肪酸和含唾液酸的寡糖鏈三部分組成。神經節苷脂鞘氨醇為鏈長 18 或 20 個碳的多羥基脂肪胺;脂肪酸是通過亞氨基連結在神經鞘氨醇基上,主要是硬脂酸(18:0)占 95%,其他 5% 的脂肪酸有蛛網酸(20:0)、棕櫚酸(16:0)或棕櫚油酸(16:1)。脂肪酸和鞘氨醇組成神經酰胺(ceramide),是神經節苷脂結構中的疏水部分。神經節苷脂中的寡糖鏈主要為 D-葡萄糖(D-Glc)、D-半乳糖(D-Gal)、D-乙酰半乳糖胺(GalNAc)。寡糖鏈末端的唾液酸以乙酰化形式存在,稱為 N-乙酰神經氨酸(NeuNAc,NANA)。寡糖鏈和唾液酸上的多羥基組成神經節苷脂的親水部分[2-3]。因此,兩性分子神經節苷脂是以微泡或聚合物形式溶于水。
1.2 生物學功能
神經節苷脂廣泛存在于哺乳動物的組織和細胞膜上,其中在腦組織和神經組織中含量最高。作為膜抗原和病毒、細菌及毒素的受體,GLS 在細胞識別,細胞粘合,調節細胞免疫,決定血型等方面起著非常重要的作用[4-6]。神經節苷脂水解產生的神經酰胺(ceramide),是一種抑制性第二信使,參與多種細胞功能,具有調節細胞生長、變異、凋亡,調節蛋白分泌,參與免疫過程與炎癥反應等功能[7]。其中含有一個唾液酸的 GLS 稱為單唾液酸四己糖神經節苷脂,它能夠通過血-腦脊液屏障,聚集受損腦區嵌入細胞膜,模仿內源性 GM1發揮作用,能夠促進神經細胞生存、軸突生長和突觸生長的神經重構,恢復由于各種原因引起的中樞神經系統損傷的功能,而且對損傷后繼發性神經退化和腦水腫也有積極作用[8-11]。1996 年我國從阿根廷 TRB Pharma 公司引進了利用豬腦生產的 GM1注射液,商品名為 Sygen(施捷因),用于治療顱腦損傷、神經退化、帕金森綜合征等腦部疾病。
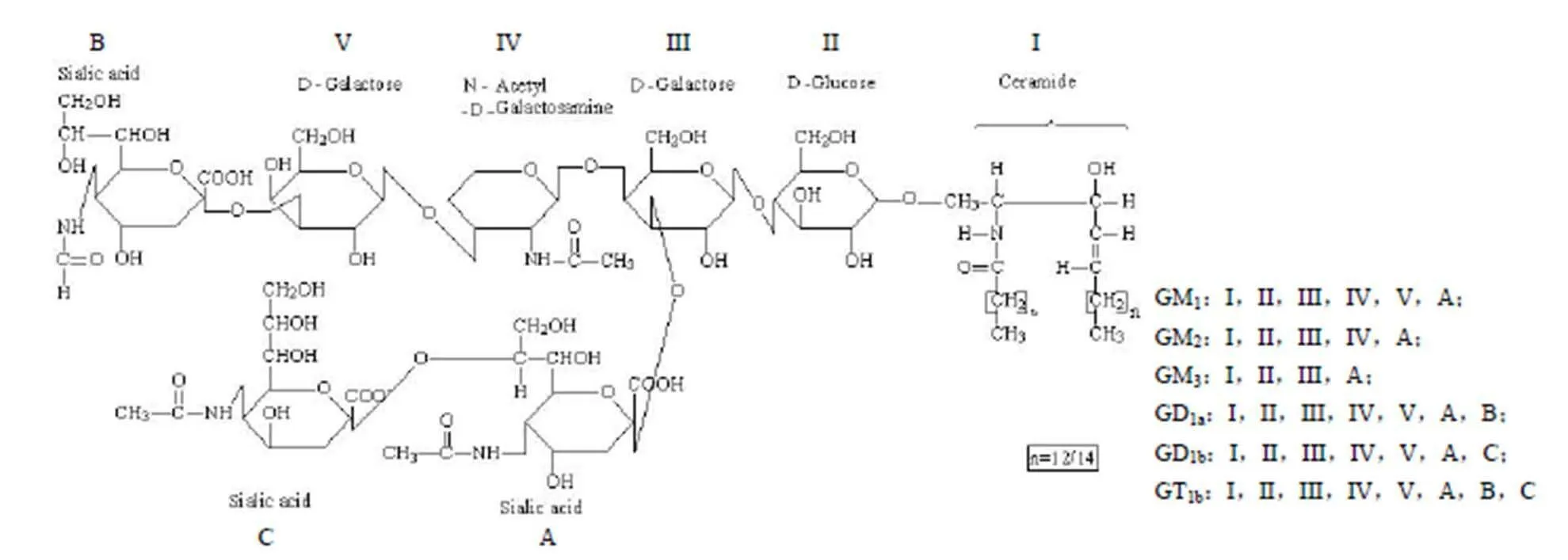
圖 1 神經節苷脂結構[12]
國內齊魯制藥有限公司生產的相同規格的豬腦來源 GM1鈉注射液(商品名:申捷)于 2004 年上市,獲得了良好的臨床應用效果。
2 唾液酸酶
2.1 唾液酸酶類型
唾液酸酶(sialidase)又稱神經氨酸酶(neuraminidase),是水解酶系中的大家族,按水解方式可分為內切唾液酸酶(exo-α-sialidase,EC 3.2.1.18)和外切唾液酸酶(endo-α- sialidase,EC 3.2.1.129);按照來源可分為哺乳動物唾液酸酶(共有 Neu1、Neu2、Neu3、Neu4 4個類型)、細菌唾液酸酶和流感病毒唾液酸酶(N1 ~ N9 共 9 個亞型)。序列分析表明細菌唾液酸酶的大小從 40 到 115 kDa不等,唾液酸酶蛋白質序列中除了兩處保守的模塊 FRIP(Phe-Arg-Ile- Pro)和 Asp box(Ser/Thr-X-Asp-[X]-Gly-X-Thr-Trp/Phe)外,其余序列并不保守。由致病菌或病毒分泌的唾液酸酶是四個結構完全相同的單體亞基組合而成的四聚體,唾液酸酶基因由基因編碼[13-15],因物種不同長度從 1 200 ~ 1500 bp不等。Roggentin 等[16]首次從中成功克隆了唾液酸酶基因,為研究唾液酸酶的結構和功能提供了便利。盡管微生物唾液酸酶的蛋白序列并不保守,但它們的空間結構具有保守的典型特征(圖2),主要是 β 螺旋功能域、免疫球蛋白樣模塊和半乳糖結合功能域(卷曲構型)[17]。需特別指出的是唾液酸酶是流感病毒的抗原決定簇之一,隨著病毒的不斷復制和變異,唾液酸酶會發生氨基酸突變,形成不同的亞型,目前發現有 9 個亞型。目前已知共有 70 多個物種的基因組中含有唾液酸酶基因,編碼 137 種不同類型的唾液酸酶,包括人、靈長類動物、海洋軟體動物、原蟲和各種微生物,其中非致病性普通微生物有糖多孢紅霉菌()、灰色鏈霉菌()、谷氨酸棒桿菌()、阿維鏈霉菌()、煙曲霉()、禾谷鐮孢菌()、樹干畢赤酵母()、游海假交替單胞菌()、煙草節桿菌()等。有的土壤微生物綠色小單胞菌()分泌的唾液酸酶有兩種形式,一種僅含有水解功能域,大小約為 42 kDa,另一種則含有水解功能域和半乳糖結合功能域,大小約為 68 kDa,前一種形式的唾液酸酶與流感病毒分泌的唾液酸酶相似,出現不同大小唾液酸酶是由不同的培養基中不同碳源誘導所致,但具體的誘導物和誘導機制還不清楚[18]。
2.2 唾液酸酶功能、性質和活性測定
在流感病毒或致病菌中,唾液酸酶能夠將病毒顆粒表面含唾液酸的糖蛋白進行水解,使病毒顆粒脫離宿主細胞進行傳播和擴散。而在一些非致病菌如土壤細菌中,唾液酸酶主要的功能則是水解唾液酸用作碳源或能源進行物質代謝[19]。唾液酸酶在弱酸性條件下(pH 5.0 ~ 5.5)活性最強,而且受多種無機離子的影響,其中鹵素離子 Cl–和 Br–能夠顯著提高唾液酸酶活性[20],2 mmol/L 濃度的 Hg2+、Cu2+、Fe2+則會抑制唾液酸酶的活性[21]。唾液酸酶酶活定量測定有多種方法,基本原理是利用神經氨酸衍生物作為底物,經唾液酸酶酶解后釋放衍生部分作為待測定物質,利用顏色反應或熒光檢測來測定唾液酸酶活性,主要方法有 4-甲基傘形酮基法、溴吲哚基法和苯酮苷法[22-23]。Yao 等[24]則利用離子交換色譜和酸性茚三酮測定唾液酸酶活性。經典的硫巴比妥酸法(TBA)則是測定唾液酸酶水解唾液酸衍生物后的唾液酸含量來確定,NANA 被過碘酸鈉氧化生成 β-醛基丙酮酸,繼而與硫代巴比妥酸偶聯生成紅色化合物,該化合物在 549 nm 波長下有最大吸收[25]。
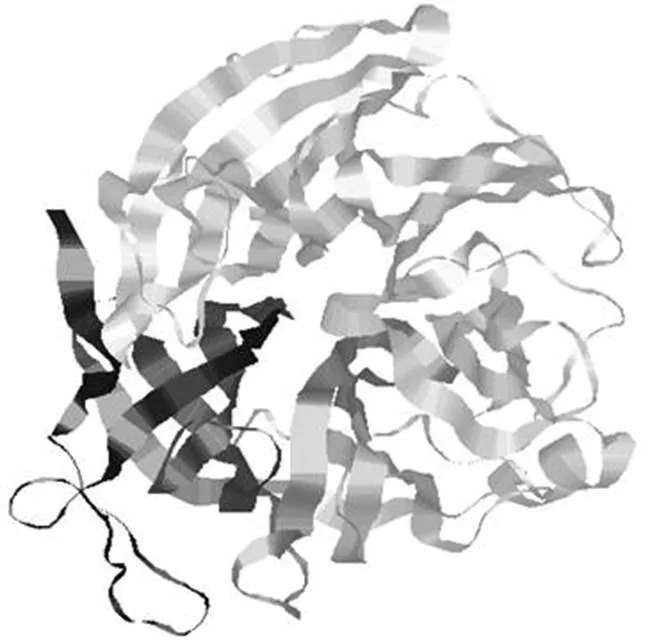
圖 2 唾液酸酶蛋白結構
3 微生物酶法轉化神經節苷脂
3.1 轉化機制
從神經節苷脂的糖鏈結構上可以看出有三種水解酶可以破壞寡糖鏈,分別為神經節苷脂內切酶、外切半乳糖甘酶和唾液酸酶,但事實上只有唾液酸酶適用于水解神經節苷脂,而不破壞 GM1中唾液酸。唾液酸酶負責催化唾液酸與糖酯之間糖苷鍵中酮基的水解,唾液酸酶不同于神經節苷脂內切酶,后者水解切割神經酰胺與寡糖鏈間的糖苷鍵。微生物來源唾液酸酶能夠水解 α-2, 3、α-2, 6 和 α-2, 8 糖苷鍵,但對 α-2, 3 糖苷鍵的水解活性高于 α-2, 6 和 α-2, 8 糖苷鍵,而且微生物唾液酸酶對于處于寡糖鏈非末端的 α-2, 3 糖苷鍵連接的唾液酸沒有水解活性[26],利用這一特性可以將三唾液酸神經節苷脂和雙唾液酸神經節苷脂水解成單唾液酸四己糖神經節苷脂,而 GM1則不會被脫去唾液酸(表1)。
3.2 應用
GLS 在動物腦組織中含量豐富,在腦灰質中含量較高,例如豬腦和牛腦,其中豬腦 GLS的總含量為豬全腦濕重的 0.0894%,高于牛腦的 0.0536%,而且牛腦提取物容易被病毒污染,所以豬腦是理想的 GM1來源。豬腦 GLS 的主要成分有 GM1、GM2、GD1a、GD1b和 GT1b[27]。目前其生產工藝大多采用溶劑萃取、酸法水解轉化和柱層析等分離提取程序從腦組織中制備 GM1單體。該方法制備 GM1需要消耗大量有機溶劑,成本高,而且剩余大量的多唾液酸神經節苷脂無法應用,是一種很大的浪費。因此,利用微生物或唾液酸酶進行神經節苷脂轉化能夠提高神經節苷脂轉化效率,提高 GM1的收率。目前國內外一些學者對微生物法轉化神經節苷脂進行了研究,并成功應用于 GM1的規模化生產中,取得了良好效果。

表 1 唾液酸酶對不同神經節苷脂的水解位點和活性

表 2 酸法和微生物酶法轉化神經節苷脂比較
華東理工大學王學東等[28-29]利用能夠分泌唾液酸酶的乳酪短桿菌()進行神經節苷脂的微生物轉化,考察了 pH、溫度、轉化體積、底物濃度對轉化的影響。結果表明乳酪短桿菌能夠分泌特異性水解末端 α-2, 3和 α-2, 8 糖苷鍵的唾液酸酶,可將神經節苷脂中的 GD 和 GT 組分有效地轉化為 GM1,使粗脂中 GM1的含量從 9% 提高至 45%,而且該酶并不催化水解 GM1中的唾液酸。日本學者 Fukano 和 Ito[30]從海洋微生物中篩選到一株產唾液酸酶的菌株 YF-2,經鑒定為假單胞菌屬()。YF-2 菌株分泌的唾液酸酶大小為 110 kDa,也具有神經節苷脂 GD1a、GD1b和 GT 的底物特異性,對 GM1沒有水解活性。25 ℃條件下培養 YF-2 菌株 3 d 可將 80% ~ 90% 的神經節苷脂轉化為 GM1。除了上述已經應用于神經節苷脂轉化的微生物外,綠色小單胞菌也能夠高效分泌唾液酸酶,重組綠色小單胞菌唾液酸酶蛋白產品已經用于臨床免疫學檢測。我們對綠色小單胞菌分泌唾液酸酶進行了研究,發現在沒有聚唾液酸或神經氨酸誘導情況下,該菌也能夠高效分泌唾液酸酶,而且具有與上述研究中微生物唾液酸酶相同的水解特性,下一步我們將用家蠶絲素凝膠固定化綠色小單胞菌制備成柱式填充床反應器對神經節苷脂進行轉化,建立適合于工業化生產的 GM1微生物轉化和制備工藝,旨在提高神經節苷脂轉化效率并實現連續化生產。
相對于酸法轉化,利用微生物和唾液酸酶轉化法生產 GM1具有專一性高,反應條件溫和等優點(表2),可以將多唾液酸神經節苷脂專一性地轉化為 GM1,提取分離后未轉化的多唾液酸神經節苷脂仍可用于下一批次的轉化,經轉化后的樣品中幾乎不含 GT 組分,僅含少量 GD 組分,轉化效果明顯優于酸轉化法,并且減少了溶劑消耗。而且微生物培養和轉化過程的成本低,能夠顯著提高生產效率,降低 GM1的生產成本。
4 結語與展望
盡管微生物酶法轉化神經節苷脂生產 GM1已經能夠成功應用于工業化生產,但是目前發現的能夠分泌唾液酸酶的微生物培養復雜,生長周期長,唾液酸酶分泌能力有待提高,這些因素直接影響了轉化神經節苷脂的效率。與酸法相比,微生物酶法轉化神經節苷脂的主要缺點就是轉化時間長。因此,可以考慮從以下幾個方面提高轉化效率:通過對現有菌株進行誘變處理,獲得高唾液酸酶活性菌株,縮短轉化時間;從分泌唾液酸酶的微生物中獲取唾液酸酶編碼基因,采用適當的高表達載體,在大腸桿菌或酵母菌中高效表達分泌型唾液酸酶,實現高效轉化;將固定化細胞技術和轉化分離耦合相結合,建立微生物轉化神經節苷脂的連續生產系統,能夠在一定時期內連續不斷的將轉化后的 GM1分離出來,從而提高轉化效率。
[1] Shioiri Y, Kurimoto A, Ako T, et al. Energy-resolved structural details obtained from gangliosides. Anal Chem, 2009, 81(1):139-145.
[2] Kato T, Hatanaka K. Purification of gangliosides by liquid-liquid partition chromatography. J Lipid Res, 2008, 49(11):2474-2478.
[3] Sonnino S, Mauri L, Chigorno V, et al. Gangliosides as components of lipid membrane domains. Glycobiology, 2007, 17(1):1R-13R.
[4] Wei JS, Fujita M, Nakai M, et al. Protective role of endogenous gangliosides for lysosomal pathology in a cellular model of synucleinopathies. Am J Pathol, 2009, 174(5):1891-1909.
[5] Miyamoto K, Takada K, Furukawa K, et al. Roles of complex gangliosides in the development of experimental autoimmune encephalomyelitis. Glycobiology, 2008, 18(5):408-413.
[6] Yanagisawa K. Role of gangliosides in Alzheimer's disease. Biochi Biophys Acta, 2007, 1768(8):1943-1951.
[7] Berenson CS, Gallery MA, Smigiera JM, et al. The role of ceramide of human macrophage gangliosides in activation of human macrophages. J Leukoc Biol, 2002, 72(3):492-502.
[8] Qiao GF, Cheng ZF, Huo R, et al. GM1 ganglioside contributes to retain the neuronal conduction and neuronal excitability in visceral and baroreceptor afferents. J Neurochem, 2008, 106(4):1637-1645.
[9] Duan JG, Xiang T, Chen H, et al. Role of extrinsic ganglioside GM1 in proliferation and differentiation of neural stem cells. J Sichuan Univ (Med Sci Edi), 2007, 38(2):260-263. (in Chinese)
段建剛, 項濤, 陳紅, 等. 外源性神經節苷脂對神經干細胞增殖、分化作用的初步研究. 四川大學學報(醫學版), 2007, 38(2):260- 263.
[10] Fighera MR, Royes LF, Furian AF, et al. GM1 ganglioside prevents seizures, Na+,K+-ATPase activity inhibition and oxidative stress induced by glutaric acid and pentylenetetrazole. Neurobiol Dis, 2006, 22(3):611-623.
[11] Chen ZG, Lu YC, Zhu C, et al. Effects of ganglioside GM1 on reduction of brain edema and amelioration of cerebral metabolism after traumatic brain injury. Chin J Traumatol, 2003, 6(1):23-27.
[12] Ariga T, Yu RK, Suzuki M, et al. Characterization of GM1ganglioside by direct inlet chemical ionization mass spectrometry. J Lipid Res, 1982, 23(3):437-442.
[13] Newstead SL, Potter JA, Wilson JC, et al. The structure of Clostridium perfringens NanI sialidase and its catalytic intermediates. J Biol Chem, 2008, 283(14):9080-9088.
[14] Chavas LM, Tringali C, Fusi P, et al. Crystal structure of the human cytosolic sialidase Neu2. Evidence for the dynamic nature of substrate recognition. J Biol Chem, 2005, 280(1):469-475.
[15] Crennell SJ, Garman EF, Laver WG, et al. Crystal structure of a bacterial sialidase (from Salmonella typhimurium LT2) shows the same fold as an influenza virus neuraminidase. Proc Natl Acad SciU S A, 1993, 90(21):9852-9856.
[16] Roggentin P, Rothe B, Lottspeich F, et al. Clonging and sequencing of a Clostridium perfringens sialidase gene. FEBS Lett, 1988, 238(1):31-34.
[17] Gaskell A, Crennell S, Taylor G. The three domains of a bacterial sialidase: a beta-propeller, an immunoglobulin module and a galactose-binding jelly-roll. Structure, 1995, 3(11):1197-1205.
[18] Watson JN, Newstead S, Narine AA, et al. Two nucleophilic mutants of the Micromonospora viridifaciens sialidase operate with retention of configuration by two different mechanisms. Chembiochem, 2005, 6(11):1999-2004.
[19] Vimr ER, Kalivoda KA, Deszo EL, et al. Diversity of microbial sialic acid metabolism. Microbiol Mol Biol Rev, 2004, 68(1):132-153.
[20] Nagaoka M,Shiraishi T,Furuhata K, et al. Effects of inorganic anions on the activation of acid sialidases. Biol Pharm Bull, 2003, 26(3):295- 298.
[21] van Aswegen CH, Dirksen van Sckalckwyk JC, du Toit PJ, et al. The effect of calcium and magnesium ions on urinary urokinase and sialidase activity. Urol Res, 1992, 20(1):41-44.
[22] Watts AG, Oppezzo P, Withers SG, et al. Structural and kinetic analysis of two covalent sialosyl-enzyme intermediates on Trypanosoma rangeli sialidase. J Biol Chem, 2006, 281(7):4149-4155.
[23] Myers RW, Lee RT, Lee YC, et al. The synthesis of 4-methylumbelliferyl alpha-ketoside of N-acetylneuraminic acid and its use in a fluorometric assay for neuraminidase. Anal Biochem, 1980, 101(1):166-174.
[24] Yao K, Ubuka T, Masuoka N, et al. Assay of sialidase activity using ion-exchange chromatography and acidic ninhydrin reaction.J Chromatogr, 1992, 581(1):11-15.
[25] Jiang ZY, Woollard AC, Wolff SP. Lipid hydroperoxide measurement by oxidation of Fe2+ in the presence of xylenol orange. Comparison with the TBA assay and an iodometric method. Lipids, 1991, 26(10): 853-856.
[26] Tiralongo J, Pegg MS, von Itzstein M. Effect of substrate aglycon on enzyme mechanism in the reaction of sialidase from influenza virus. FEBS lett, 1995, 372(2/3):148-150.
[27] Hirabayashi Y, Nakao T, Irie F, et al. Structural characterization of a novel cholinergic neuron-specificganglioside in bovine brain. J Biol Chem, 1992, 267(18):12973-12978.
[28] Wang XD, Yin Z, Peng Y, et al. Highly efficient conversion of polysialoganglioside to GM1 with Brevibacterium casei as a microbial biocatalyst. Biocatal Biotransformation, 2005, 23(1):29-32.
[29] Peng YF, Wang XD, Wei DZ. Development of a large scaleprocess for the conversion of polysialogangliosides to monosialotetrahexosylganglioside with a novel strain of Brevibacterium casei producing sialidase. Biotechnol lett, 2007, 29(6):885-889.
[30] Fukano Y, Ito M. Preparation of GM1 ganglidoside with sialidase-producing marine bacteria as a microbial biocatalyst. Appl Environ Microbiol, 1997, 63(5):1861-1865.
董惠鈞,Email:dhjjjy@gmail.com
2009-11-02
10.3969/cmba.j.issn.1673-713X.2010.02.012
