遼寧地區一脊髓小腦性共濟失調大家系癥狀前患者的基因診斷研究
2010-08-21 00:21:56曹東華劉曉莉金春蓮邱廣斌
中國實驗診斷學 2010年9期
曹東華,王 謙,劉曉莉,金春蓮,邱廣斌
(1.中國人民解放軍第二0二醫院檢驗科,遼寧沈陽 110003;2.中國醫科大學高職學院;3.中國醫科大學基礎醫學院醫學遺傳學教研室)
遼寧地區一脊髓小腦性共濟失調大家系癥狀前患者的基因診斷研究
曹東華1,3,王 謙2,劉曉莉3,金春蓮3,邱廣斌1*
(1.中國人民解放軍第二0二醫院檢驗科,遼寧沈陽 110003;2.中國醫科大學高職學院;3.中國醫科大學基礎醫學院醫學遺傳學教研室)
目的確定遼寧地區一脊髓小腦性共濟失調大家系患者子女中目前健康的癥狀前患者。方法本家系已通過基因診斷的方法明確為SCA3型,所以提取本家系患者子女外周血DNA后,采用聚合酶鏈反應對SCA3基因三核昔酸重復片段進行擴增,并將PCR異常擴增片斷測序,以明確異常擴增等位基因內重復拷貝數。結果44名患者子女中有8人SCA3基因三核苷酸重復片斷在70-82次,超過了正常人SCA3基因內三核苷酸重復的次數。結論通過基因檢測診斷出此8人為SCA3型脊髓小腦性共濟失調癥狀前患者。
脊髓小腦性共濟失調;三核苷酸重復;癥狀前患者
(Chin J Lab Diagn,2010,14:1420)
脊髓小腦性共濟失調(spinocerebellar ataxia,SCA),是一大類由于遺傳因素造成的進行性神經系統變性疾病,是一種單基因遺傳病,多為常染色體顯性遺傳。其病理損害的主要部位是脊髓、腦干、小腦[1,2],臨床上主要表現為共濟失調,構音障礙、意向性震顫以及其它小腦癥狀。由于該病起病隱匿,臨床癥狀互相重疊,基因檢查是確診和分型的唯一方法。我們于2009年在遼寧地區發現一脊髓小腦性共濟失調大家系,經家系調查和基因診斷確定該家系為脊髓小腦性共濟失調3型。由于該類疾病突變基因的相對穩定性,所以我們可以通過基因篩查發現癥狀前患者,并進行跟蹤、臨床隨訪,了解該病的發展過程。在本研究中,按照患者家屬要求,我們對家系中患者的后代進行基因檢測,現報告如下。
1 材料與方法
1.1 研究對象
本家系共5代198人(圖1),前4代每代均有患者。由于年代久遠,不能獲得本家系中第1代的詳細信息,因此不能明確致病基因來自Ⅰ1還是Ⅰ2。第2代也只能找到本身是患者且留下患者后代的分支,而第2代的正常分支則無從調查,因此本家系中第2代都是患者。本家系共有患者30人,目前存活13人。患者發病年齡最大39歲,最小20歲。前三代發病年齡都在35歲以上,第四代出現一例27歲發病患者和一例20歲發病患者。病史及體征基本相同,主要表現為行走不穩、步態蹣跚、飲水嗆咳、語音低弱、構音障礙等共濟失調癥狀和體征。我們通過基因檢測,已確定本家為SCA3型(待發表)。
1.2 研究方法
1.2.1 前提條件 按照知情同意原則,基因測試必須獲得受試者同意。向所有申請基因診斷者說明基因測試的目的、程序、可能的結果、目前能夠檢測的范圍,并保證保守秘密、尊重其隱私權,如果受試人無法理解或為歲以下未成年人,則向其監護人、父母作出解釋。
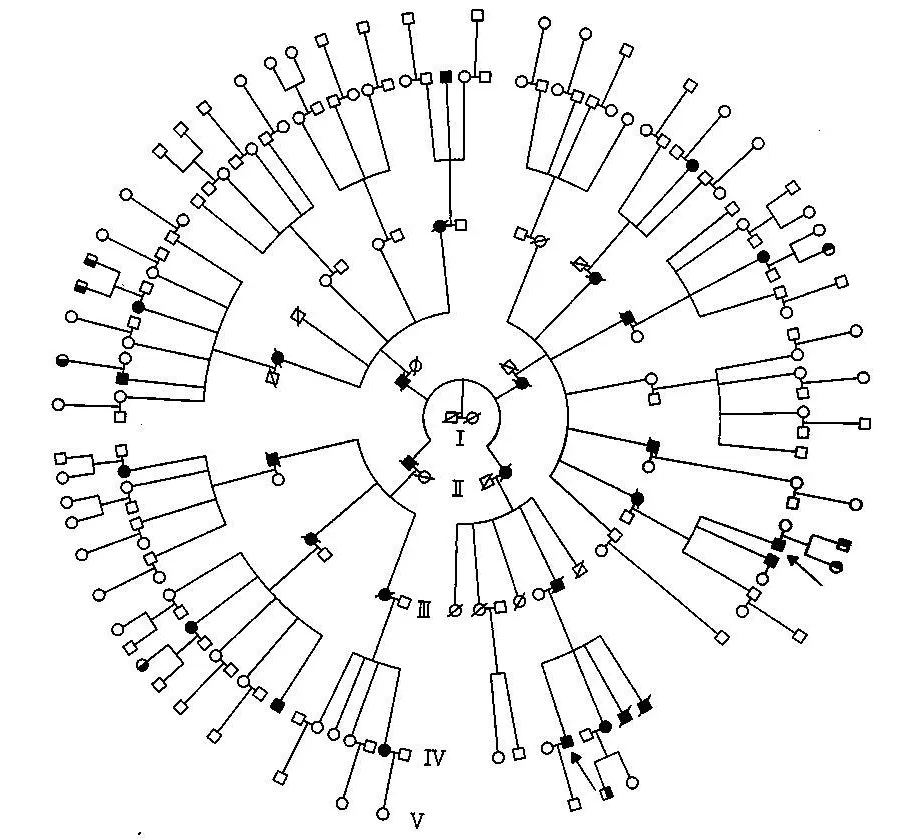
圖1 家系圖譜
1.2.2 基因組DNA提取 抽取本家系中所有患者子女中在世且未發病的成員共44人外周靜脈血4 ml,枸椽酸鈉抗凝,應用Tiangen血液DNA提取試劑盒提取基因組DNA作為模板。
1.2.3 SCA3三核苷酸重復片段PCR擴增 引物由上海生工合成,序列如下:Forward:5′-CTTTGATTCGTGAAACAATG-3′;Reverse 5′-GCCTTACCTAGATCACTCCC-3′。PCR 體系 :總體積 25 μ L,緩沖液 2.5 μ L,10×buffer 2.5 μ L,dNTP 2 μ L,上下游引物(20 μ mol/L)分別0.5 μ L,模板基因組DNA 約100 ng,TaKaLa Taq酶0.25 U,蒸餾水補齊,PCR反應在熱循環儀上進行。循環參數如下:95℃預變性5 min后,進行35個循環:95℃變性45 s,55℃退火45 s,72℃延伸45 s,最后一次72℃延伸10 min。PCR產物2%瓊脂糖凝膠電泳→發現異常條帶后將異常條帶純化回收→與pMD18-T載體連接,轉化于大腸桿菌JM109感受態細胞,經過藍白篩選→取陽性克隆搖菌→菌液送金思特公司測序。
1.2.4 遺傳咨詢 檢查結果書面通知受試者本人或其監護人,不得將測試結果告知第三者。向能夠接受基因測試并可以理解者或其監護人解釋的遺傳方式、基因突變形式、簡單的發病機理、目前的治療現狀,鼓勵增強治病信心,學習必要的康復訓練,加強鍛煉。對未成年人強調不得告知陽性結果,不得歧視,保證其順利成長。針對存在的外顯率問題,慎重解釋可能出現的后果。
2 結果
2.1 PCR產物檢測結果 本家系患者子女中在世且未發病者共44人,其中8人SCA3基因CAG三核苷酸重復片段PCR擴增后有異常等位基因(圖2)。
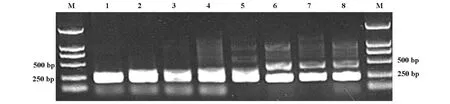
圖2 8名無癥狀患者子女SCA3基因PCR擴增產物電泳圖
2.2 異常等位基因CAG三核苷酸重復片段測序結果 8人SCA3基因 CAG三核苷酸重復70-82次(圖3),證實此8人為癥狀前患者。
3 討論
隨著分子生物學技術水平的不斷提高,醫學遺傳學、分子遺傳學的發展,基因檢查已逐漸走向臨床,人們對脊髓小腦性共濟失調這一大類疾病的認識以及發現率也不斷提高。通過基因檢測、分型有利地促進了的研究進展。按照分子遺傳學分類,迄今為止共發現30種亞型。近年來大部分亞型的基因已被定位克隆,并且明確了三核苷酸重復序列動態突變,即致病基因內三核苷酸(CAG)n的拷貝數逐代增加的突變是主要致病原因。采用基因分型可以對患者作出準確的基因診斷,也正是由于直接而可靠的基因診斷技術的發展才有癥狀前診斷的出現。遺傳性共濟失調的突變機制的揭示使得可以在任何年齡階段都能直接進行DNA檢測,這必然帶來復雜的與之相關的倫理、醫學、社會問題。
國內外對其他神經遺傳病的癥狀前診斷報道較多,但是關于SCA的癥狀前研究較少。國外學者對基因診斷操作提出了諸多相關的倫理問題并傾向于嚴格規范,并強調遺傳咨詢的重要性。癥狀前基因檢測具有高度的敏感性和個人重要性,關系受試者本人的醫療保險、應聘就業、婚姻等,因而確保隱私是幾乎所有受試者最優先考慮的問題。國內有關這方面的問題目前雖然還不十分突出,但是隨著文化素質的提高、生活水平的改善,上述問題勢必會同國外一樣變得現實,應當引起重視[3,4]。因此我們在癥狀前患者的篩查時給予當事人或其監護人準確的遺傳咨詢十分重要。在進行遺傳咨詢時除了講清SCA的遺傳方式、臨床表現、簡要發病機理及預后外,還有必要講明與測試有關的倫理、社會、法律、心理問題等[5]。
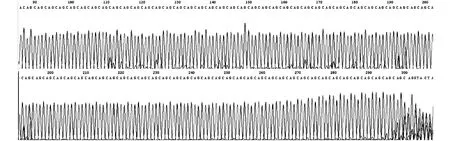
圖3 癥狀前患者SCA3基因(CAG)n三核苷酸重復片段測序圖
SCA3最早報道于葡萄牙人種,隨后的發現顯示這是一種全球性的疾病,在中國、日本及德國均為最常見的SCA類型[6],在中國的SCAs中約占一半左右。SCAs部分與三核苷酸重復序列有關,也有部分與點突變有關,還有部分未知。SCA3基因定位于14q24.3-q32.2[7],SCA3型患者是由于三核苷酸CAG重復次數增多所致,SCA3患者CAG三核苷酸重復數為53-86次,而正常人≤47次。介于47-53之間的中間類型有少量報道也顯示存在異常臨床表型[8]。本家系中在世的患者子女共44人,其中第3代4人,第4代22人,第5代16人。經基因檢測分析,只有第5代中患者子女有8人的SCA3基因CAG三核苷酸重復在此范圍內,可以推斷此8人為癥狀前患者。第3代中患者子女僅存4人,而且年齡都在50歲以上,目前還沒有發病,很可能是不攜帶致病基因,與基因檢測結果相符。第4代中患者子女未發病者共22人,經基因檢測此22人都不攜帶致病基因。此22人中大部分都在35歲以上,基本超過了本家系發病年齡,另有兩人未到30歲,1人32歲,也不攜帶致病基因。第5代中患者子女共16人,年齡最大22歲,最小1歲,經基因檢測,有8人攜帶致病基因。
有的遺傳性疾病如肝豆狀核變性,通過基因檢測后發現癥狀前患者,給予一定的藥物治病,可以預防該疾病的發生。但SCA缺乏有效的治療手段,Miller等[9]報道可用RNA干擾的方法使基因沉默,但應用于臨床尚有很長的距離。干細胞移植雖然可以暫時緩解SCA的癥狀,但并不能根治,而且醫療費用巨大,多數家庭承受不起。目前最為有效的控制SCA發生的手段就是應用產前診斷技術杜絕可能發病的后代出生。我們準備在此家系患者和癥狀前患者本人或愛人懷孕時,動員孕婦抽取羊水,以鑒定胎兒是否攜帶致病基因。如果胎兒攜帶致病基因則及時人工流產,以避免此家系再次出現患者,減輕其家族和社會的負擔。
[1]Duenas A M,Goold R,Giunti P.Molecular pathogenesis of spinocerebellar ataxias[J].Brain,2006,129:1357.
[2]Netravathi M,Pal PK,Purushottam M,et al.Spinocerebellar ataxias types 1,2 and 3:age adjusted clinical severity of disease at presentation correlates with size of CAG repeat lengths[J].J Neurol Sci,2009,277(1-2):83.
[3]謝秋幼,李詢樺,梁秀齡,等.脊髓小腦性共濟失調的癥狀前診斷研究[J].腦與神經疾病雜志,2004,12(30):193.
[4]張小寧,雷 晶,馬建華,等.脊髓小腦性共濟失調癥狀前診斷的初步研究[J].腦與神經疾病雜志,2007,15(2):95.
[5]Tan EK,Ashizawa T.Genetic testing in spinocerebellar ataxia:defining a clinical role[J].Arch Neurol,2001,58(2):191.
[6]姜曉華,葉 蕾,傅 毅,等.遺傳性脊髓小腦型共濟失調一例家系SCA3基因突變研究[J].中華醫學雜志,2005,85(12):848.
[7]謝秋幼,梁秀齡,李洵樺,等.我國南方漢族人脊髓小腦性共濟失調不同基因亞型的頻率分布[J].中華檢驗醫學雜志,2004,27(9):555.
[8]van Alfen N,Sinke RJ,ZwartsMJ,et al.Intermediate CAG repeat lengths(53,54)forMJD/SCA3 are associated with an abnormal phenotype[J].Ann Neurol,2001,49:805.
[9]Miller V,Xia H,Marrs G,et al.Allele-specific silencing ofdominant disease genes[J].PNAS,2003,100:7195.
Gene diagnosis of asymptomatic carriers in a big spinocerebellar ataxias pedigree in LiaoNing
CAO Dong-hua,WANGQian,LIU Xiao-li,et al.(Department of Test,No.202Hospital of People'sLiberation Army,Shenyang110003,China)
ObjectiveTo identify the asymptomatic carrierswhose parents are affected in a big spinocerebellar ataxiaspedigree.Methods44 blood sampleswere collected from descendants of the patients in this Pedigree whichwas confirmed as SCA3 in previous study.The fragments of the SCA3 gene containing the CAG repeat region were amplified by means of polymerase chain reaction(PCR).The number of CAG repeatsin abnormal allele fragmentswas identifiedby using agarose gel electrophoresis(AGE)and DNA sequencing.ResultsThe CAG repeatnumbers in eight sampleswere in the range of 70-82,whichexceededthe normal range.ConclusionThe eight samples are SCA3 asymptomatic carriers by gene diagnosis.
Spinocerebellar ataxia;Trinucleotide repeat;Asymptomatic carriers
R745.1
A
1007-4287(2010)09-1420-03
*通訊作者
2010-02-08)
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
海峽科技與產業(2016年3期)2016-05-17 04:32:12
