
喘立停氣霧劑中麻黃堿提取和純化工藝的研究
2010-09-09 08:09:48周丹英余琪錢冬偉朱琦
中成藥 2010年6期
關鍵詞:工藝
周丹英,余琪,錢冬偉,朱琦
(浙江省中藥研究所,浙江杭州 310023)
喘立停氣霧劑中麻黃堿提取和純化工藝的研究
周丹英,余琪,錢冬偉,朱琦
(浙江省中藥研究所,浙江杭州 310023)
麻黃;正交試驗;麻黃總堿;提取率;有機溶劑;萃取
目的:研究從麻黃草中提取麻黃堿的工藝;選擇適宜的萃取溶劑、萃取次數等,以得到較高純度的麻黃堿。方法:采用正交試驗法進行麻黃的提取工藝優選,利用高效液相色譜法測定麻黃堿的含量。結果:在研究的幾個因素中,提取次數對提取率有顯著性影響,對麻黃堿含量有較大影響。結論:最佳提取工藝為藥材8倍量水回流提取,提取2次,每次提取2 h;純化工藝則選擇用乙醚作為萃取溶劑,用與堿化后麻黃浸膏等體積的乙醚萃取3次,回收乙醚即可得到麻黃總堿浸膏,其純度達到85%以上。該方法提取率高,準確穩定,易于操作。
麻黃為麻黃科植物草麻黃Ephedra sinicaStapf、中麻黃Ephedra intermediaSchrenk et C.A.Mey.或木賊麻黃Ephedra equisetina Bag.的干燥草質莖[1]。味辛、微苦,性溫,具發汗散寒、宣肺平喘、利水消腫之功效。其主要化學成分為麻黃堿、偽麻黃堿。
“喘立停”處方為臨床使用多年的經驗方,將麻黃、細辛、椒目、艾葉等藥材合理組合配伍使用,用于治療哮喘,控制哮喘發作時的癥狀,具有良好的臨床效果。作為平喘用藥,其作用的主要靶部位是肺,而吸入療法是哮喘治療的首選方法之一[2],可避免肝臟及胃腸道的首過作用,吸收后迅速進入體循環,作用快,劑量小,副作用小[3],因此我們選擇氣霧劑進行本新藥的研究。本論文主要研究麻黃藥材中麻黃總堿的提取和純化工藝研究,以確定最佳麻黃堿提取工藝和純化工藝。
為探討麻黃提取工藝的科學性,我們以麻黃總堿、提取率為指標,采用水加熱提取,正交試驗優選其提取工藝的最佳條件。同時,建立了麻黃堿的含量測定方法,并進行了方法學考察。
1 儀器與試藥
1.1 儀器Agilent 1100高效液相色譜儀(安捷倫公司);DH9140-3鼓風電熱恒溫干燥箱(上海精密實驗儀器設備有限公司);senco r-501-旋轉蒸發儀(上海申順生物科技有限公司);lambda20紫外分光光度儀(PE):AB204-5電子天平(Mettler Tolldo); KQ100-DB數控超聲清洗器(昆山市超聲儀器有限公司)。
1.2 試藥鹽酸麻黃堿對照品、鹽酸偽麻黃堿對照品(中國藥品生物制品檢定所,批號分別為17124-200303,171237-200304)。麻黃藥材,購于亳州新興中藥材飲片有限公司,經鑒定為麻黃科植物草麻黃Ephedra sinicaStapf的干燥草質莖;甲醇(分析純)、乙腈(色譜純)。
2 實驗方法和結果
2.1 麻黃總堿含量測定方法[4]
2.1.1 色譜條件選擇以十八烷基鍵合硅膠為填充劑;色譜柱C18(4.6 mm×250 mm,5 μm);流動相:乙腈-0.2%磷酸溶液(4∶96),流速1.0 mL/ min,柱溫30℃;檢測波長205 nm。外標法測定麻黃堿含量。
2.1.2 對照品溶液的制備精密稱取真空干燥至恒重的鹽酸麻黃堿對照品和鹽酸偽麻黃堿對照品適量,加流動相溶解,制成每1 mL約含150 μg鹽酸麻黃堿和50 μg鹽酸偽麻黃堿的混合溶液,作為對照品溶液。
2.1.3 線性關系考察精密稱取鹽酸麻黃堿和鹽酸偽麻黃堿,置50 mL量瓶中,加流動相溶解并定容,得到每1 mL含0.148 8 mg鹽酸麻黃堿和0.048 mg鹽酸偽麻黃堿,按上述色譜條件進樣量分別為2、4、6、8、10、20、30 μL測定。以峰面積為橫坐標(X),鹽酸麻黃堿進樣量為縱坐標(Y),進行回歸,得回歸方程Y=0.007 7X+0.007 2(R=0.999 9)。結果表明,鹽酸麻黃堿在0.297 6~4.464 μg范圍內與峰面積呈良好的線性關系。以峰面積為橫坐標(X),鹽酸偽麻黃堿進樣量為縱坐標(Y),得回歸方程為Y=0.000 81X+0.003 4(R=0.999 9)。結果顯示鹽酸偽麻黃堿在0.096~1.44 μg范圍內與峰面積呈良好的線性關系。
2.1.4 精密度試驗精密吸取上述對照品溶液,重復進樣5次,測定峰面積,鹽酸麻黃堿平均峰面積為3 865.2,RSD為0.06%(n=5),鹽酸偽麻黃堿平均峰面積為1 191.6,RSD為1.349%。
2.1.5 重復性試驗按擬定的方法對同一樣品進行6次平行供試品溶液制備與測定。結果鹽酸麻黃堿平均含量RSD=1.49%(n=6),鹽酸偽麻黃堿RSD=2.97%(n=6)。
2.1.6 加樣回收試驗精密稱取已知含量的供試品,分別精密加入鹽酸麻黃堿和鹽酸偽麻黃堿的混合對照品(濃度分別為0.80 mg/mL、0.29 mg/mL)1 mL,制成供試品溶液,依法進行測定,計算。鹽酸麻黃堿平均回收率為98.78%,RSD為1.95%;鹽酸偽麻黃堿平均回收率為98.77%,RSD為1.64%。
2.1.7 供試品溶液的制備、測定取正交試驗樣品1~9號,精密吸取樣品溶液1.0 mL,置25 mL量瓶中,加流動相[乙腈-0.2%磷酸溶液(4∶96)]稀釋至刻度,超聲提取20 min,取出,搖勻后用0.45 μm的微孔濾膜濾過,取續濾液即得。
2.2 提取率的測定[5]精密吸取樣品溶液10 mL,置已干燥至恒重的蒸發皿中,將其水浴蒸干后,置于同一恒溫箱中,105℃干燥3 h,取出后置干燥器內冷卻30 min,分別精密稱重,計算提取率。
2.3 麻黃堿的提取
2.3.1 提取溶媒的確定麻黃堿屬苯異丙胺衍生物,可溶于水、乙醇、乙醚等溶劑中,因此可采用水提、醇提、醚提等方法。目前醫院制劑室提取麻黃生物堿方法有3種,即水蒸汽蒸餾法、酸水溫浸法、酸水煮提法[6]。作者對酸水、水、稀乙醇提取麻黃總堿作了比較,按常規提取條件進行:每份取100 g麻黃藥材,提取2次、每次2 h,每次用10倍量提取溶媒,結果見表1。
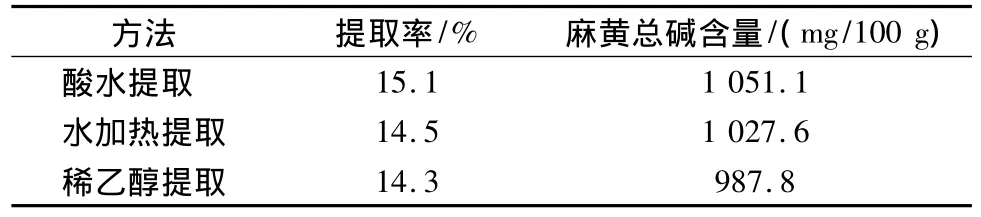
表1 幾種方法提取麻黃的比較
各方法所得麻黃堿含量和提取率差異不是非常明顯,我們從對設備的要求和操作安全性考慮,認為還是選擇用水加熱提取法比較合適。
2.3.2 實驗方法分別稱取麻黃100 g 9份,按正交試驗表選擇的條件進行提取,合并提取液,過濾,旋轉蒸發減壓濃縮至一定體積V,作為樣品溶液。
2.3.3 試驗安排選擇提取次數、提取時間、溶媒用量3個因素,按下表進行L9(34)正交試驗。
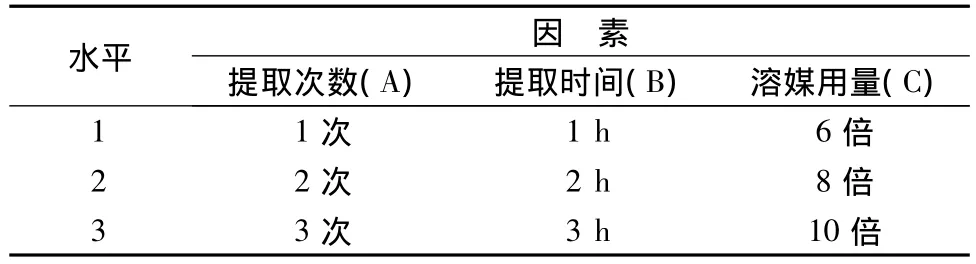
表2 試驗因素水平表
2.3.4 正交試驗結果與分析按以下正交試驗安排表進行實驗,麻黃總堿和提取率測定結果見表3。
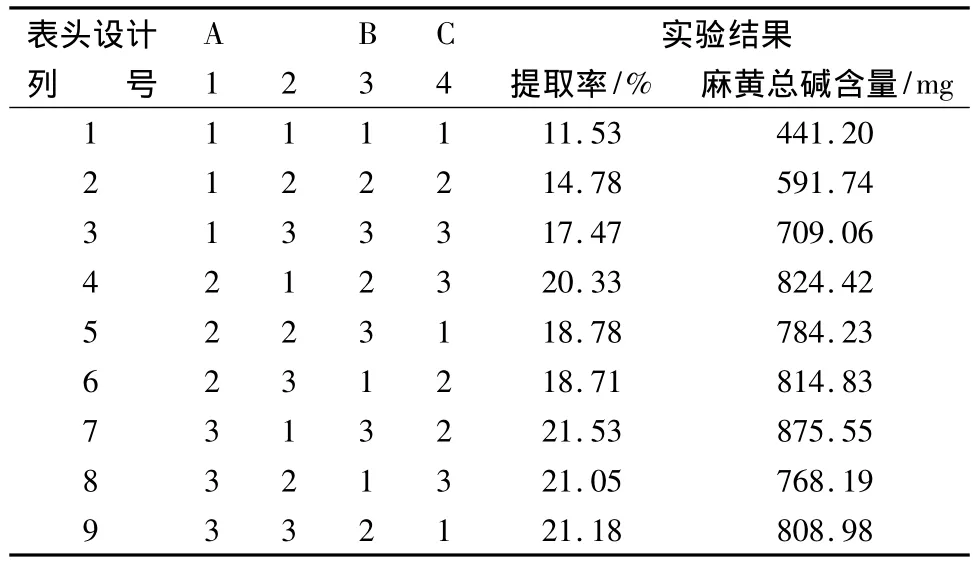
表3 L9(34)正交試驗設計表試驗數據
分別對有關實驗數據作極差和方差分析,結果見表4和表5。A因素對提取率有顯著性影響,對麻黃總堿含量也有較大影響。以上各因素對提取率來說影響程度依次為A>C>B,而對麻黃總堿來說依次為A>B>C。根據實驗結果和從省時節能、降低成本的角度考慮,優選出最佳提取工藝為A2B2C2,即麻黃的提取工藝確定為:麻黃藥材8倍量水提取2次,每次提取2 h。
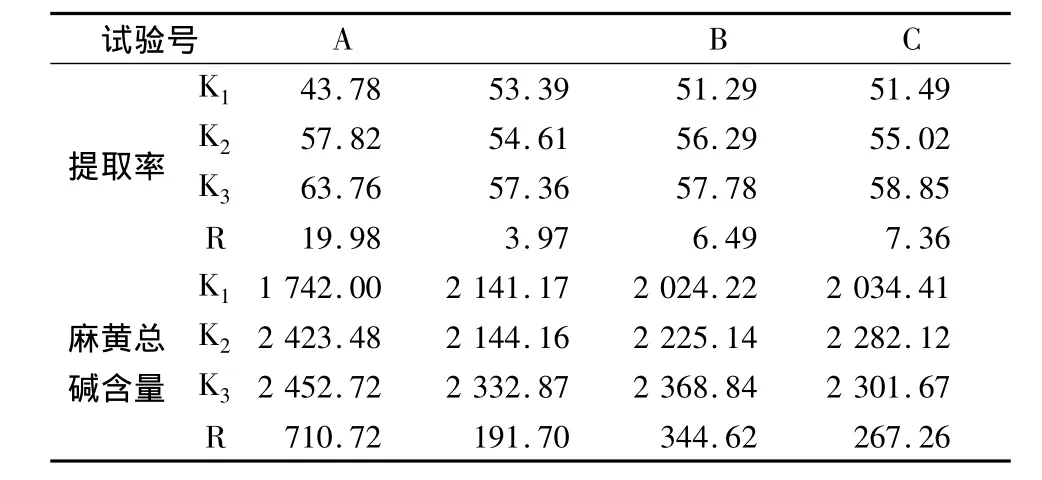
表4 極差分析
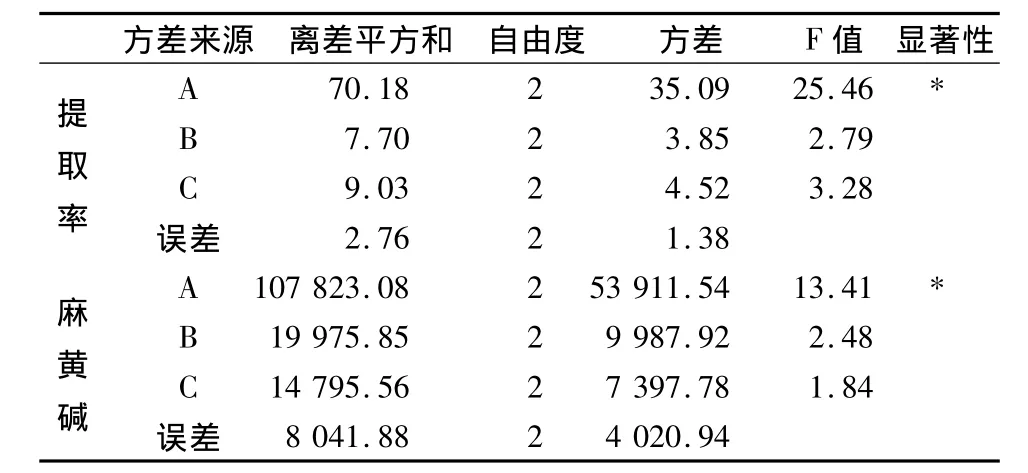
表5 方差分析
2.3.5正交驗證試驗稱取100 g麻黃藥材各3份,按上述優選出的提取條件進行試驗,分別測定各樣品的提取率(%)和麻黃總堿總量(mg)。結果見表6。
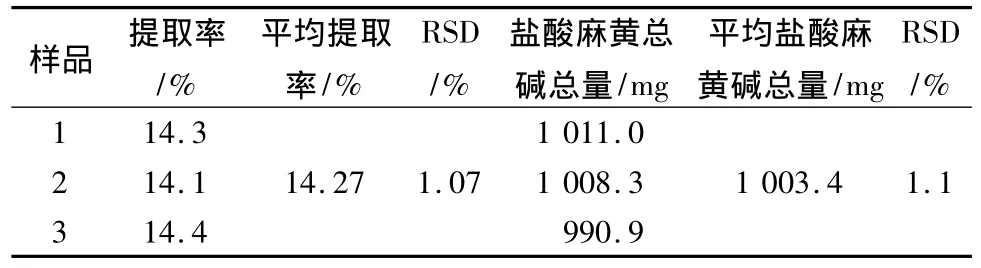
表6 正交驗證試驗結果
結果表明,按正交試驗優選出的提取條件切實可行,重現性良好。
2.4 麻黃總堿的精制和純化
2.4.1 麻黃提取液中麻黃總堿萃取溶劑的確定麻黃經提取、濃縮后得到的是麻黃浸膏(密度為1.1左右),而喘立停作為氣霧劑則需要得到較高純度的麻黃堿,因此必須進行麻黃堿的純化和精制。根據麻黃堿的性質我們曾采用了甲苯和苯、氯仿、乙醚等有機溶劑進行萃取。因考慮到部分有機溶劑操作上的安全性和藥用的安全性,最后選擇無水乙醚作為溶劑。2005版藥典中乙醚是二級溶劑,因此我們做了麻黃堿中乙醚殘留量測定。
2.4.2 麻黃總堿萃取次數的確定取出304 g麻黃浸膏,測得麻黃總堿含量為20.66 mg/g,加入20%NaOH溶液調節pH至12,分別加堿化后藥液等體積的無水乙醚萃取4次,分別測定每次乙醚萃取液中鹽酸麻黃總堿含量。經檢測分析:第1次萃取液中含5 556 mg麻黃總堿,第2次萃取液含741 mg,第3次萃取液含364 mg,第4次萃取液中已經檢測不到麻黃總堿。
結果:麻黃藥液用乙醚萃取3次,鹽酸麻黃總堿回收率可達97.67%。因此選擇每次用與堿化后浸膏等體積的乙醚萃取3次,萃取液回收乙醚后置水浴上蒸干可得到較高純度的麻黃總堿。
2.5 麻黃總堿中乙醚殘留量測定
2.5.1 方法照氣相色譜法(中國藥典2005年版一部附錄ⅥE.)測定。
色譜條件與系統適應性試驗色譜柱:HP-5(5%苯基甲基硅酮)(30 m×320 μm×0.25 μm);柱溫:起始為35℃,維持4 min,再以30℃/min升至180℃,維持5 min,再以30℃/min降至35℃,維持2 min;進樣口溫度:200℃;檢測器溫度:250℃;載氣:高純氮;分流比50∶1;檢測器:FID(空氣流速450 mL/min,氫氣流速45 mL/min);進樣量:1 μL。
對照品溶液的制備精密稱取乙醚適量,加二甲基甲酰胺溶解,制成每1 mL約含0.5 mg乙醚的溶液,作為對照品溶液。
供試品溶液的制備精密取本品0.1 g,加二甲基甲酰胺溶解定容至5 mL,作為供試品溶液。
測定法分別精密吸取對照品溶液與供試品溶液各1 μL,注入氣相色譜儀,測定,即得。
2.5.2 樣品測定數據見表7。

表73 批麻黃堿樣品乙醚含量(n=3)
從上表得知,無水乙醚萃取后得到的麻黃堿浸膏經檢測沒有乙醚殘留。
綜上所述,麻黃提取、純化工藝確定為:麻黃藥材8倍量水提取2次,每次2 h,提取的藥液經減壓濃縮得到比重為1.10左右的麻黃藥液。用與堿化后的麻黃藥液等體積的乙醚萃取3次,所得萃取液經乙醚回收后置水浴上蒸干即可得到高純度的麻黃總堿,其純度可達85%以上。
3 討論
3.1 提取溶媒的選擇經預試驗表明,采用水加熱回流提取麻黃堿能得到比較好的提取效果。因此從設備安全性和生產成本考慮還是選擇水加熱回流提取。
3.2 麻黃堿的測定采用高效液相色譜法,經方法學考察實驗表明,本方法線性好,精密度高,重現性等都較好,測得結果準確。
3.3 提取率作為中藥制劑的重要考察指標;中藥制劑是粗中取精,最大程度地將藥材中有效成分提取出來,因此將提取率和麻黃總堿含量作為關鍵性指標。
3.4 無水乙醚萃取麻黃堿得率高,因乙醚為二級溶劑,因此特地進行了乙醚殘留量測定,經氣相檢測麻黃總堿中沒有乙醚殘留。這與乙醚本身物理性狀密切相關,乙醚極易揮發,萃取液經回收乙醚后置水浴上蒸干至恒重,即可以得到較高濃度的麻黃堿浸膏(深褐色稠膏)。
[1]中國藥典[S].一部.2005:223.
[2]李明華,殷凱生,朱栓立.哮喘病學[M].北京:人民衛生出版社,1998:345.
[3]畢殿洲.藥劑學[M].4版,北京:人民衛生出版社,1999:388.
[4]中國藥典[S].一部.2005:附錄56.
[5]劉貴中.高效液相色譜法測定鼻炎滴劑中鹽酸麻黃堿的含量[J].藥物分析雜志,1998,18(5):341.
[6]嚴曉梁,鄧曉鴻.幾種實驗室提取麻黃堿方法的比較[J].華西藥學雜志,1994,9(4):265-267.
R284.2
A
1001-1528(2010)06-0950-03
2009-04-15
浙江省科技廳科研院所研究開發專項(2005F13006)
周丹英(1976-),女,工程師,主要從事中藥研究。Tel:(0571)85229698 E-mail:zhoudy1976@163.com
猜你喜歡
中國特種設備安全(2022年5期)2022-08-26 09:19:32
礦產綜合利用(2020年1期)2020-07-24 08:50:40
山東冶金(2019年6期)2020-01-06 07:45:54
收藏界(2019年2期)2019-10-12 08:26:06
世界農藥(2019年2期)2019-07-13 05:55:12
世界農藥(2019年2期)2019-07-13 05:55:10
模具制造(2019年3期)2019-06-06 02:11:00
山東工業技術(2016年15期)2016-12-01 05:30:59
銅業工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
