
負載型非晶態Co-B催化劑在1-辛烯氫甲酰化中的應用
2010-11-06 07:01:45龍俊英賀德華
物理化學學報 2010年10期
關鍵詞:催化劑
龍俊英 馬 蘭,2 賀德華,*
(1清華大學化學系,有機光電子與分子工程教育部重點實驗室,分子催化與定向轉化研究室,北京 100084;2防化指揮工程學院,北京 102205)
負載型非晶態Co-B催化劑在1-辛烯氫甲酰化中的應用
龍俊英1馬 蘭1,2賀德華1,*
(1清華大學化學系,有機光電子與分子工程教育部重點實驗室,分子催化與定向轉化研究室,北京 100084;2防化指揮工程學院,北京 102205)
以醇凝膠-氮氣熱處理方法制備的ZrO2、碳納米管(CNTs)及介孔分子篩SBA-15作為載體,采用化學還原法制備了負載型非晶態Co-B催化劑,利用粉末X射線衍射(XRD)、氮氣吸附等溫線、透射電子顯微鏡(TEM)、電感耦合等離子體-原子發射光譜(ICP-AES)以及X射線光電子能譜(XPS)等對催化劑進行了表征.以1-辛烯的氫甲酰化為目標反應,考察了負載型非晶態Co-B催化劑在氫甲酰化反應中的性能及循環使用效果.表征結果顯示,以NaBH4為還原劑制備的負載型Co-B催化劑上的Co-B為非晶態,負載Co-B組分對載體的晶相結構沒有影響.反應結果表明,在1-辛烯的氫甲酰化反應中,負載型非晶態Co-B催化劑都顯示較高的初始活性.隨著循環次數的增加,三種催化劑的活性下降,但下降幅度有所不同,Co-B/CNTs和Co-B/SBA-15的循環使用穩定性高于Co-B/ZrO2.
Co-B非晶態催化劑; 負載; 氫甲酰化; 催化性能
α-烯烴氫甲酰化可以得到比初始烯烴反應物多一個碳原子的醛(或進一步氫化得到醇)[1].氫甲酰化的反應產物被廣泛用作溶劑、交聯劑或洗滌劑的初始原料[2-4].目前,工業上氫甲酰化反應使用的商業催化劑主要是銠配合物[5-6]和鈷配合物催化劑[7],例如著名的Wilkinson催化劑RhCl(PPh3)3(Ph=phenyl).均相催化過程有著它的優勢,例如獨特的活性位點,易于與配體連接從而調變催化性能,溫和的反應條件,高催化活性和選擇性.然而,和所有的均相催化過程相似,工業氫甲酰化反應也面臨著催化劑和產物的分離問題.因此,氫甲酰化催化劑的非均相化是一項非常有意義的工作.現今已有不少關于氫甲酰化均相過程多相化的研究,如使用水溶性催化劑[8]、兩相催化劑[9]、固載型配合物催化劑[10-12]和固體催化劑[13]等.傳統負載型鈷和銠金屬催化劑應用于氫甲酰化研究也取得了一定的進展[14-15],但是催化劑的活性、選擇性和穩定性仍需要進一步提高.另一方面,開發新型的氫甲酰化催化材料也是引人關注的熱點之一.1980年以來,有關非晶態合金的催化性能研究受到了人們的關注,但主要集中在Ni-B、Cu-B催化劑的制備及其在烯烴、炔烴和硝基化合物加氫反應中的應用[16-18].非晶態合金催化劑由于其長程無序而短程有序的特殊結構和性能,使得非晶態合金可望成為新型催化材料之一[19].非負載型Co-B非晶態催化劑從1980年以來一直被用于醛加氫或者是烯烴加氫反應[20-22].近來我們研究了Co-B非晶態催化劑(非負載型)在氫甲酰化反應中的應用,表明Co-B非晶態催化劑對氫甲酰化反應具有良好的催化性能[23].但由于非晶態催化劑本身比表面積較小和熱穩定性較差,人們開始嘗試將非晶態催化劑Ni-B、Co-B負載在具有較大比表面積的載體上,并取得較好的效果[24-25].本文對負載型Co-B催化劑在氫甲酰化反應中的催化性能進行研究,選用介孔分子篩SBA-15、碳納米管(CNTs)以及ZrO2作為載體,采用化學還原法制備了負載型Co-B/ZrO2、Co-B/CNTs和Co-B/SBA-15非晶態合金催化劑,表征了它們的物理化學性質,并選擇1-辛烯氫甲酰化為目標反應,考察其在氫甲酰化反應中的催化活性、選擇性和催化劑循環使用的穩定性.初步討論了催化劑在循環使用中活性下降的原因.
1 實驗部分
1.1 實驗藥品
1-辛烯購自美國 ACROS公司,純度 99%; CNTs由清華大學反應工程研究所提供;CoCl2· 6H2O購自廣東汕頭市西隴化工廠,分析純;NaBH4、ZrOCl2·8H2O、甲苯和無水乙醇均購自北京化工廠,均為分析純.
1.2 催化劑的制備
CNTs用濃HNO3處理并干燥[26]后使用,ZrO2采用沉淀-醇凝膠-N2中焙燒的方法制備[27],介孔分子篩SBA-15按照文獻[28]的方法制備,負載型Co-B催化劑采用化學還原法制備.以Co-B/ZrO2為例,將0.l mol·L-1的氯化鈷水溶液作為浸漬液,在室溫下浸漬ZrO2粉末,Co與ZrO2按1∶4(質量比)投料,攪拌下浸漬24 h后,蒸干水分,后在馬弗爐中N2氣氛下(30 mL·min-1)將其轉移至三口燒瓶中,加入去離子水調成漿態液,將三口燒瓶置于冰水浴中,在攪拌及氮氣保護下將1.0 mol·L-1的NaBH4逐滴加入到懸濁液中(NaBH4的用量為CoCl2投料量(摩爾)的2倍),滴加完后繼續攪拌直到無氣泡生成,然后再攪拌0.5 h,把固體過濾后反復水洗至中性后再用乙醇洗滌4次,在乙醇中保存,并置于充入氮氣的干燥器中.按上述同樣的方法制備Co-B/CNTs和Co-B/ SBA-15.此外,非負載型Co-B樣品按文獻方法[20]制備.
1.3 催化劑的表征
樣品的比表面積、累積孔容和平均孔徑分布的測定使用美國Micromeritics公司的ASAP-2010C型吸附儀,采用低溫(-196℃)氮氣吸附法測定.樣品測定之前先在200℃下脫氣2 h以上.樣品的比表面積采用BET法計算;平均孔體積以吸附氣體的相對壓力為0.995時的吸附量來測定;平均孔徑的計算采用BJH法并以吸附等溫線的脫附支為基準.
樣品的晶相結構使用德國 Brüker公司的D8 Advance型X射線衍射儀測定,使用CuKα射線,Ni濾色片,管電壓為40 kV,管電流為40 mA,掃描范圍為15°-80°.小角X射線衍射分析采用日本理學Rigaku公司的D/Max-2200X衍射儀,掃描范圍為0.5°-8°.
樣品的形貌通過帶有能量色散X射線能譜(EDS)分析功能的日本JEOL公司的JEM-2010高分辨透射電子顯微鏡進行表征,該透射電鏡加速電壓為120 kV.催化劑上Co、B的組成和負載量采用美國Jarrel-Asm公司Atom Scan2000型號的電感耦合等離子體-原子發射光譜(ICP-AES)測定.利用日本ULVAC-PHI Inc.公司的PHI Quantera SXM型號的X射線光電子能譜(XPS)測定催化劑Co、B表面的電子態.
1.4 催化性能測試
采用高壓反應釜對催化劑進行性能評價.反應釜內容積為100 mL,催化劑用量為0.25 g,1-辛烯為5 mL,溶劑甲苯為15 mL.反應條件為 120℃,5 MPa合成氣(H2/CO的體積比為1∶1),反應時間為150 min.將催化劑、1-辛烯、甲苯、攪拌子置入反應釜,密閉反應釜蓋后,置于冰水浴中(防止泄壓時反應物1-辛烯的揮發),用2 MPa合成氣置換釜內空氣,反復三次,泄至常壓.然后將反應釜置于加熱爐中,程序升溫至所需反應溫度,充入合成氣至所需的反應壓力,開始反應時打開電磁攪拌計.反應停止后待反應釜降至室溫,把反應釜置于冰水浴中,緩慢放氣,打開釜蓋,將反應產物倒入離心管,離心分離催化劑,取上層清液進行分析.對于催化劑的循環,離心后所得的固體干燥后用于循環使用,不再加入新鮮的催化劑.反應液用魯南SP-6890型氣相色譜儀分析氫甲酰化產物各組分的含量并計算轉化率和選擇性.GC分析條件為:HP-5毛細管色譜柱(0.25 mm×30 m),氫火焰離子化檢測器,載氣為氮氣,柱溫50-250℃(15℃·min-1),用峰面積歸一化法確定含量.
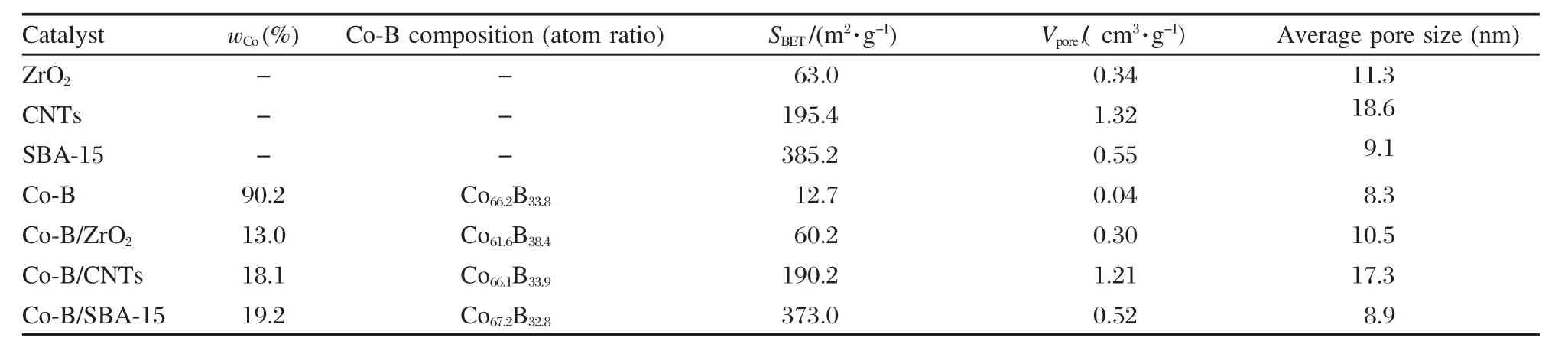
表1 載體及Co-B催化劑的組成及織構性質Table 1 Composition and texture of the supports and supported Co-B catalysts
2 結果與討論
2.1 催化劑的表征結果
載體和Co-B催化劑的比表面積等織構性質以及催化劑上Co負載量、Co-B組成等結果列于表1.由表1可以看出,載體ZrO2的比表面積較小,而CNTs和SBA-15的比表面積很大,CNTs的孔容及平均孔徑也較大.Co-B/SBA-15的比表面積為373.0 m2·g-1,Co-B/CNTs(190.2 m2·g-1)的居中,遠高于Co-B/ZrO2的比表面積.各載體負載Co-B后,比表面積有所下降,但下降不大;與非負載型Co-B的比表面積[23]相比較,負載型Co-B催化劑有較高的比表面積.不同載體對負載型Co-B催化劑上的Co/B原子比例有所影響,但Co-B/SBA-15和Co-B/CNTs上的Co/B原子比基本相近.在相同條件下制備的三種負載型Co-B催化劑的鈷的實際負載量有一定的差異,這可能與載體的比表面積大小和孔道結構有關.
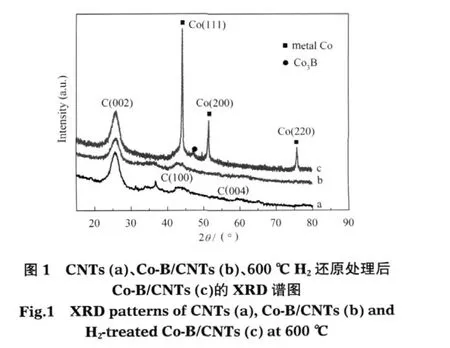
載體CNTs、新鮮Co-B/CNTs樣品以及Co-B/ CNTs用H2還原后樣品的XRD譜如圖1所示.載體CNTs的XRD譜(圖1b)中出現了碳納米管的C(002)晶面特征衍射峰,另外在2θ≈44°和2θ≈58°附近分別出現了C(100)和C(004)晶面的弱彌散峰,這與文獻[29]的報導一致.負載Co-B后的新鮮Co-B/CNTs催化劑的XRD譜(圖1b)與載體CNTs的XRD (圖1a)譜基本相同,也觀測到C(002)晶面衍射峰以及C(100)和C(004)晶面的弱彌散峰,但比較載體CNTs和Co-B/CNTs的XRD譜發現,Co-B/CNTs催化劑在2θ≈44°-45°附近的峰更寬泛,這可能是由于CNTs的C(100)晶面衍射峰與Co-B非晶態合金所具有的44°-45°附近寬化彌散特征衍射峰[20]相疊加所致.把新鮮的Co-B/CNTs在H2氣氛中600℃下還原處理后,Co-B/CNTs-H2-600樣品的XRD譜(圖1c)上,在2θ≈45°,52°,76°分別出現了金屬Co的(111)、(200)和(220)晶面的強且尖銳的衍射峰,在2θ≈47°出現了Co3B晶相的弱衍射峰.比較圖1的(b)和(c)譜圖可知,新鮮Co-B/CNTs催化劑上的Co-B為非晶態結構.
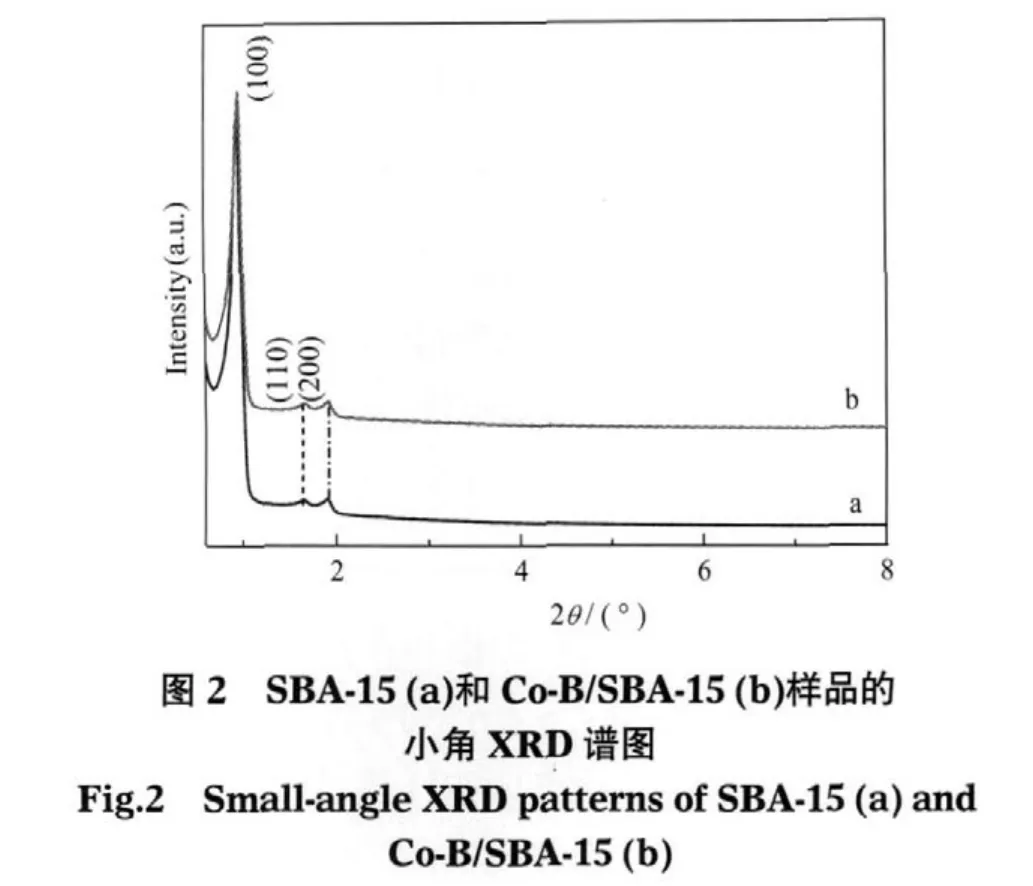
載體SBA-15和Co-B/SBA-15樣品的小角度的XRD譜如圖2所示,SBA-15浸漬催化劑后得到的Co-B/SBA-15保持了載體SBA-15材料的規則六方相結構,在2θ≈0.8°左右存在一個具有六方介孔結構的(100)面的特征衍射峰[30],表明Co-B/SBA-15仍保持SBA-15原有的規整孔道結構.
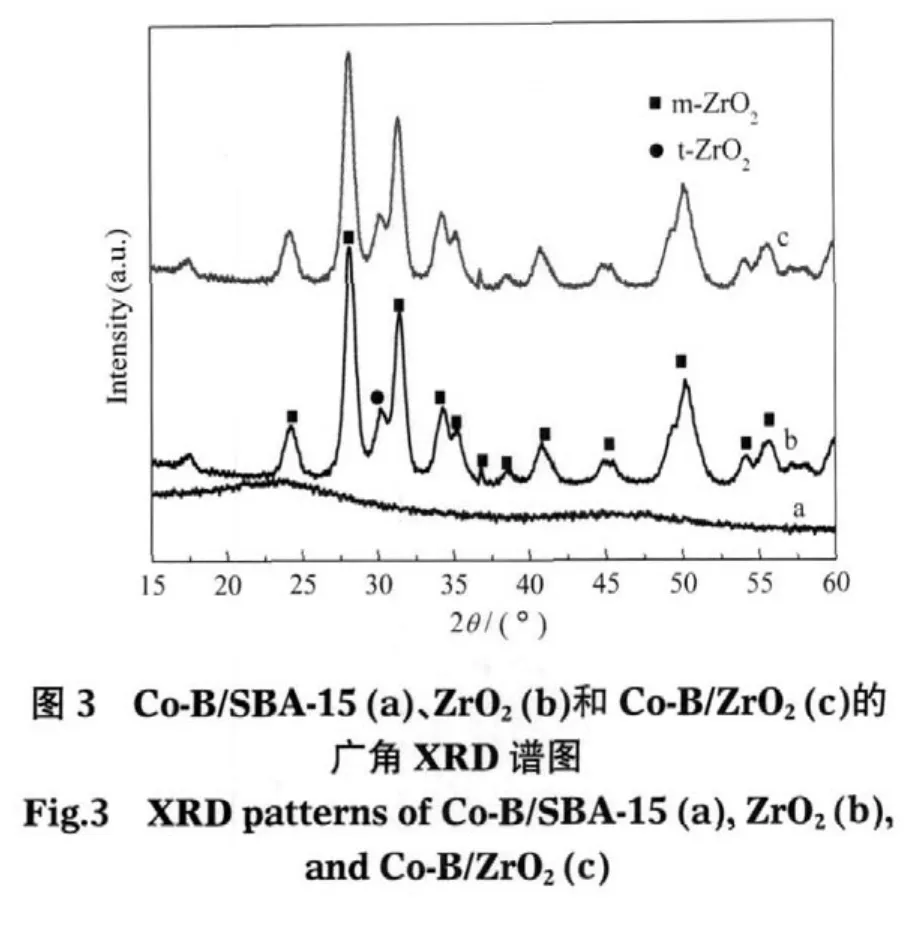
此外,Co-B/SBA-15樣品的廣角度XRD譜如圖3(a)所示.2θ≈23°處的峰是載體SBA-15產生的彌散峰,而在2θ≈45°處出現一個弱的寬化彌散峰,則歸屬于Co-B非晶態結構[31].Co-B/ZrO2的XRD譜如圖3(c)所示,與載體ZrO2的XRD譜(圖3(b))沒有大的差異,由于載體ZrO2本身(單斜晶相)在2θ≈15°-60°之間有多個衍射峰,因此負載于ZrO2上的Co-B非晶態寬化彌散峰未能明顯觀測到.
圖4是負載型Co-B催化劑的透射電鏡照片和相應的EDS圖.由圖4(a-1,a-2)可以看出,Co-B/ ZrO2催化劑顆粒較為均勻,在EDS譜分析中檢測到Co元素的信號,表明Co已經固載于ZrO2載體上.由于B的原子量小,EDS分析未檢測到B元素的信號.
從圖4(b-1)的照片可以清晰地看到載體CNTs的形貌和含Co元素的顆粒,EDS分析證明了Co元素在CNTs載體上的存在(圖4(b-2)).從圖4(c-1)照片也可以清晰看出載體SBA-15的規整孔道結構和含Co元素的顆粒,同樣EDS(圖4(c-2))結果表明Co元素在載體SBA-15表面上.
用XPS對非負載型Co-B表面的電子狀態進行了表征.圖5是Co-B的XPS譜圖.結果表明,通過NaBH4化學還原法得到的非負載型Co-B樣品上,除了有還原態的Co0(電子結合能EB(Co2p3/2)=777.8 eV)之外,還存在氧化態的 Co2+(EB(Co 2p3/2)=780.9 eV).另一方面,對于B元素,樣品表面上有還原態的B0(B 1s的電子結合能EB=187.6 eV)存在,也有氧化態的B3+(EB=191.6 eV).
2.2 負載型Co-B催化劑在1-辛烯氫甲酰化反應中的性能及循環使用效果
表2是負載型Co-B催化劑在1-辛烯氫甲酰化反應中的性能以及與非負載型Co-B催化劑性能的比較.從表2可以看出,Co-B/ZrO2、Co-B/CNTs及Co-B/SBA-15催化劑的初活性(新鮮催化劑第一次反應時的轉化率)都比較高;與非負載型的Co-B催化劑比較,負載型催化劑的1-辛烯轉化率都比非負載型Co-B的高,說明通過負載的方法可以使Co-B組分得到較好的分散,從而提高轉化率.此外,Co-B/ CNTs的初活性(轉化率為92.2%)要高于Co-B/SBA-15和Co-B/ZrO2的初活性,這可能是由于CNTs載體本身具有良好的儲氫性能[32-33],對氫的吸附有促進作用,從而對催化劑活性的提高有一定的益處.另一方面,在Co-B/CNTs及Co-B/SBA-15催化劑上,目的產物壬醛的選擇性都很高(分別為96.2%和98.7%).
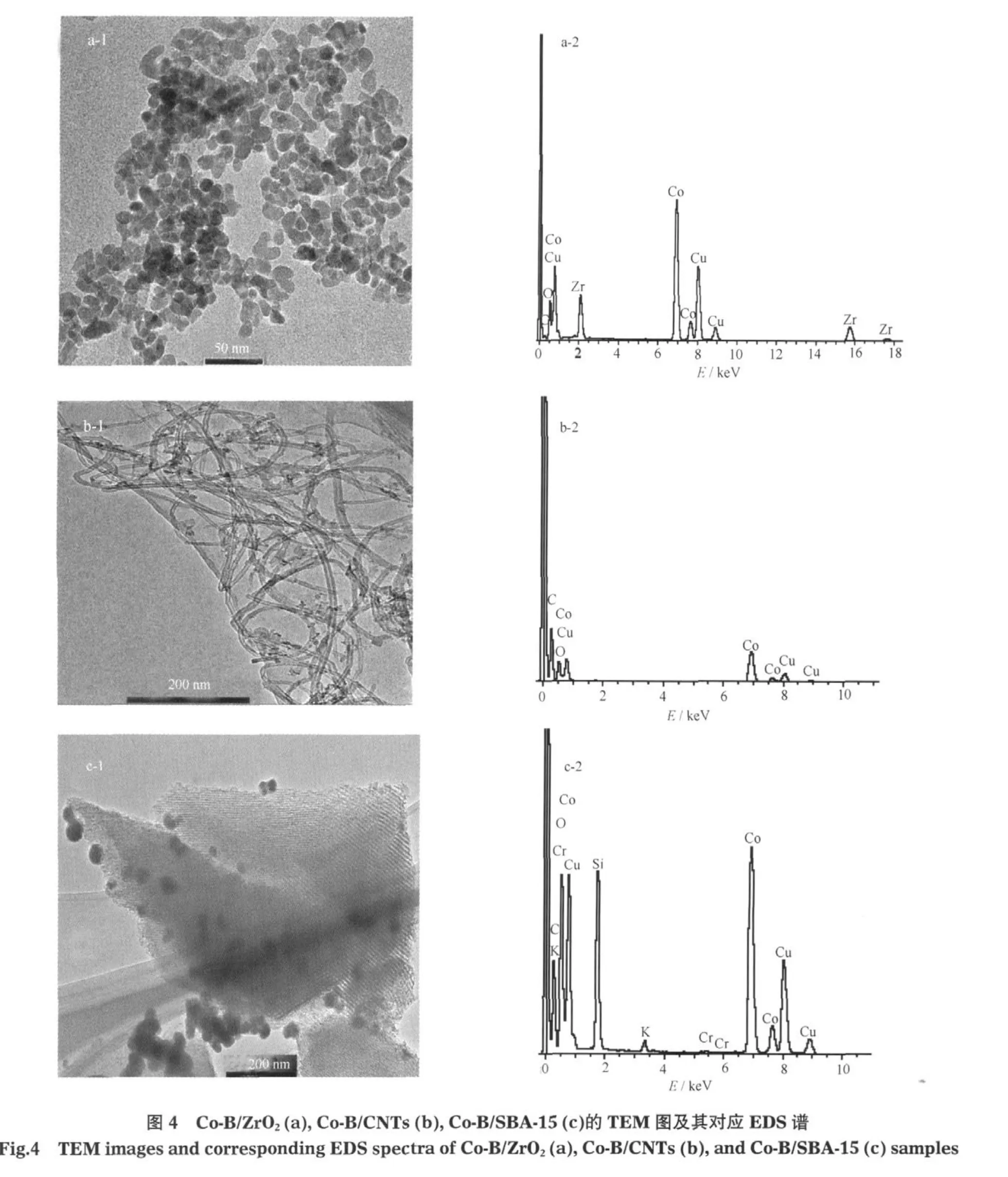
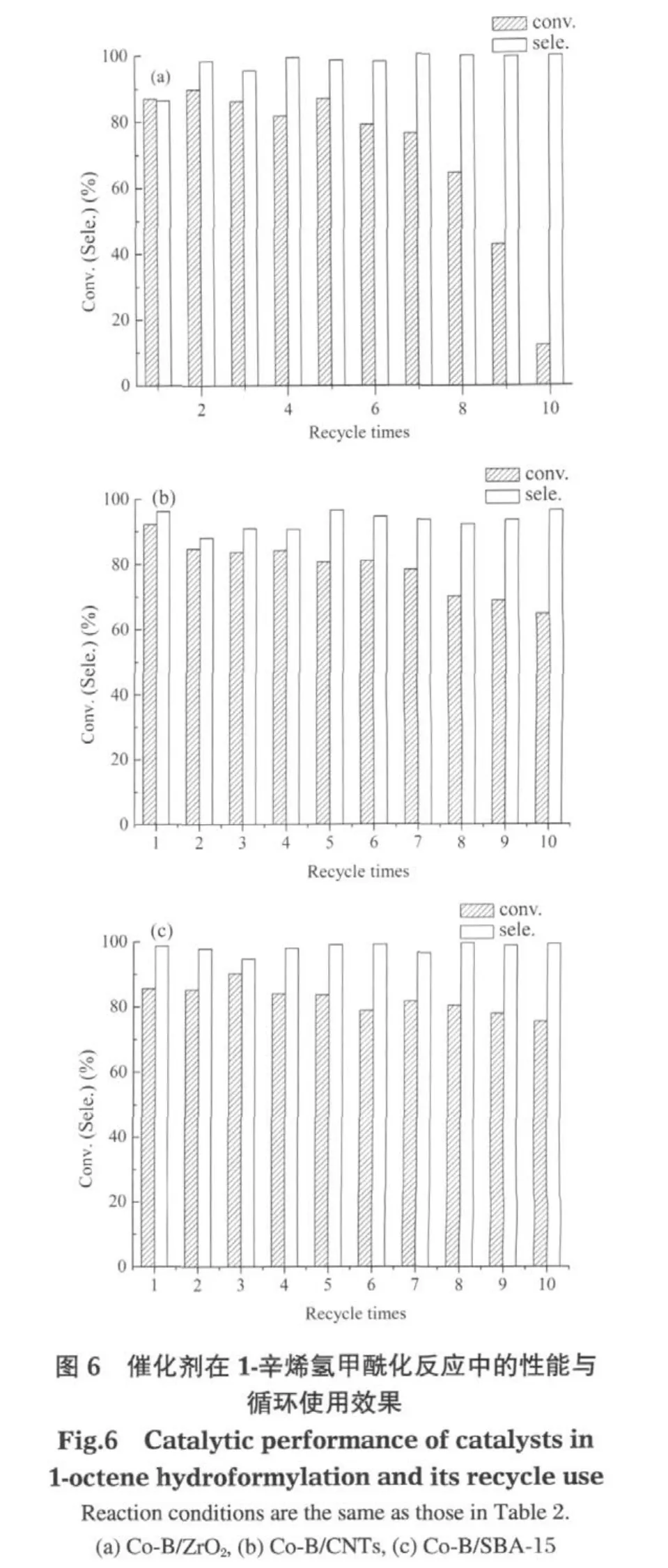
圖6是Co-B/ZrO2、Co-B/CNTs及Co-B/SBA-15三種催化劑在1-辛烯氫甲酰化反應中的循環使用效果.可以得出,隨著催化劑循環次數的增加,三種催化劑的活性都呈下降趨勢,但下降的幅度不同.在10次的循環使用過程中,Co-B/ZrO2催化劑的活性下降幅度最大,轉化率由初始反應的86.9%降到了第10次反應的12.2%;Co-B/SBA-15催化劑的活性由初始反應的 85.6%降到了第 10次反應的75.4%;Co-B/CNTs催化劑的活性由初始反應的92.2%降到了第10次反應的64.7%.Co-B/SBA-15及Co-B/CNTs在10次循環使用中活性下降幅度比Co-B/ZrO2要小得多,這可能是由于 SBA-15和CNTs都有比ZrO2高得多的比表面積的緣故,這使得Co-B在SBA-15及CNTs上分散得較好并有利于Co-B與載體較牢固地結合,使得Co-B不容易從載體上流失;此外,SBA-15表面豐富的羥基也有助于Co-B較牢固的負載.另外,對于這三種負載型催化劑,隨著循環次數的增加,壬醛的選擇性沒有大的變化.
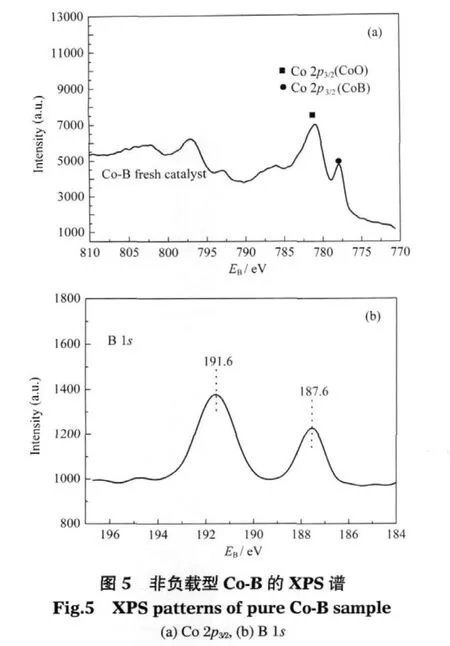
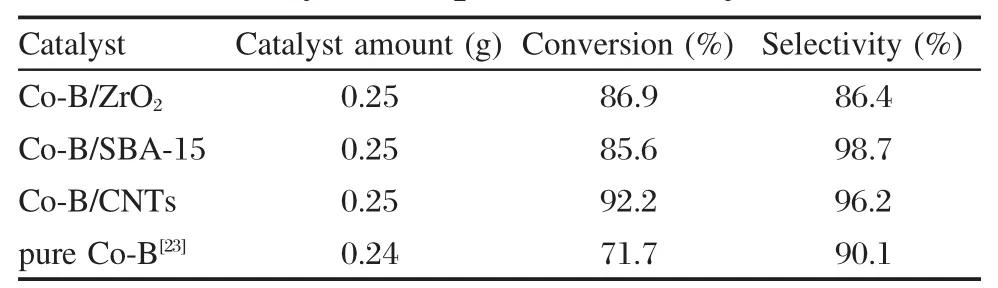
表2 負載型Co-B催化劑與非負載型Co-B催化劑的性能Table 2 Catalytic performance of supported Co-B catalysts and pure Co-B catalyst
通過ICP分析發現,催化劑在反應中Co活性組分從載體上脫落、流失于反應母液中,這是催化劑在循環使用中活性下降的原因之一.三種催化劑在循環使用中Co的流失量不一樣,分析反應后母液中的Co含量,發現Co-B/ZrO2催化劑的Co流失量最多,10次反應后累積Co流失量達到89%,而Co-B/CNTs及Co-B/SBA-15的Co流失量相對較低,分別為20%和13%.Co-B/ZrO2催化劑上Co的大量流失,是造成Co-B/ZrO2催化劑在循環使用中活性大幅下降的主要原因.對于負載型Co-B催化劑,強化Co-B組分與載體的牢固結合,防止活性組分流失,是負載型Co-B催化劑循環使用的關鍵.這方面的研究正在繼續進行之中.
3 結 論
用化學還原法制備了負載型Co-B/ZrO2、Co-B/ CNTs和Co-B/SBA-15催化劑,Co-B以非晶態形態負載于載體上,固載Co-B后載體的結構沒有改變.三種負載型非晶態Co-B催化劑在1-辛烯的氫甲酰化反應中顯示出良好的初始活性,隨著催化劑循環次數的增加,活性有不同程度的下降,其中Co-B/ ZrO2的活性下降幅度最大.反應中催化劑上Co的流失是造成催化劑在循環使用中活性下降的主要原因.
致謝: 感謝清華大學化工系反應工程研究所提供CNTs樣品,感謝清華大學分析基金的資助.
1 Cornils,B.;Rasch,M.;Otto,R.Angew.Chem.Int.Edit.,1994,33: 2144
2 Chapuis,C.;Jacoby,D.Appl.Catal.A,2001,221:93
3 Botteghi,C.;Paganelli,S.;Moratti,F.J.Mol.Catal.A-Chem., 2003,200:147
4 Russell,M.J.H.Platinum Met.Rev.,1988,32(4):179
5 Ungvary,F.C.Chem.Rev.,1997,167:233
6 Klingler,R.J.;Chen,M.J.;Rathke,J.W.;Kramarz,K.W. Organometallics,2007,26(2):352
7 Dieguez,M.;Pamies,O.;Claver,C.Tetrahedron:Asymmetry, 2004,15(14):2113
8 Lin,Q.;Jiang,W.D.;Fu,H.Y.;Chen,H.;Li,X.J.Appl.Catal.A, 2007,328(1):83
9 Fu,H.Y.;Li,M.;Chen,H.;Ji,X.J.Mol.Catal.A-Chem.,2006, 259(1-2):156
10 Zhou,W.;He,D.H.Green Chem.,2009,11:1146
11 Zhou,W.;He,D.H.Catal.Lett.,2009,127:437
12 Fierro,J.L.G.;Merchan,M.D.;Rojas,S.;Terreros,P.J.Mol. Catal.A-Chem.,2001,166(2):255
13 Kainulainen,T.A.;Nieme,M.K.;Krause,A.O.I.J.Mol.Catal. A-Chem.,1997,122:39
14 Kainulainen,T.A.;Niemela,M.K.;Krause,A.O.I.J.Mol.Catal. A-Chem.,1999,140:173
15 Zhang,H.;Qiu,J.;Liang,C.;Li,Z.;Wang,X.;Wang,Y.;Feng,Z.; Lin,C.Catal.Lett.,2005,101:211
16 Wang,M.H.;Li,H.X.;Deng,J.F.Chin.J.Catal.,1999,20:548 [王明輝,李和興,鄧景發.催化學報,1999,20:548]
17 Li,H.;Xu,Y.;Yang,H.X.;Zhang,F.;Li,H.X.J.Mol.Catal.AChem.,2009,307:105
18 Li,H.;Zhao,Q.F.;Li,H.X.J.Mol.Catal.A-Chem.,2008,285: 29
19 Lü,Z.G.;Guo,Z.M.Chem.Ind.Eng.Prog.,1998,21(8):552 [呂志果,郭振美.化工進展,1998,21(8):552]
20 Nitta,Y.;Imanaka,T.;Teranishi,S.Bull.Chem.Soc.Jpn.,1980, 53:3154
21 Chen,Y.Z.;Wu,K.J.Appl.Catal.,1991,78:185
22 Pei,Y.;Guo,P.J.;Qiao,M.H.;Li,H.X.;Wei,S.Q.;He,H.Y.; Fan,K.N.J.Catal.,2007,248:303
23 Ma,L.;Peng,Q.R.;He,D.H.Catal.Lett.,2009,130:137
24 Li,H.X.;Wang,W.J.;Li,H.;Deng,J.F.J.Catal.,2000,194: 211
25 Chen,X.Y.;Wang,S.;Zhuang,J.H.;Qiao,M.H.;Fan,K.N.;He, H.Y.J.Catal.,2004,227:419
26 Zhang,Q.;Zhao,M.Q.;Liu,Y.;Cao,A.Y.;Qian,W.Z.;Lu,Y. F.;Wei,F.Chinese Science Bulletin,2009,54:1794 [張 強,趙夢強,劉 毅,曹安源,騫偉中,盧云峰,魏 飛.科學通報, 2009,54:1794]
27 Xu,B.Q.;Wei,J.M.;Yu,Y.J.Phys.Chem.B,2003,107:5203
28 Zhao,D.Y.;Feng,J.L.;Huo,Q.S.;Melosh,N.;Fredrickson,G. H.;Chmelka,B.F.;Stucky,G.D.Science,1998,279:548
29 Ocampo,A.L.;Miranda,H.M.;Morgado,J.;Montoya,J.A.; Sebastian,P.J.J.Power Sources,2006,160(2):915
30 Wang,Y.;Noguchi,M.;Takahashi,Y.;Ohtsuka,Y.Catal.Today, 2001,68:3
31 Wonterghem,J.V.;Morup,S.C.;Koch,J.W.;Charles,S.W.; Wells,S.Nature,1986,322:622
32 Dillon,A.C.;Jones,K.M.;Bekkedahl,T.A.Nature,1997,386: 377
33 Manuela,V.;Fabrizio,C.Chem.Phys.Lett.,2003,371:476
1-Octene Hydroformylation Using Supported Amorphous Co-B Catalysts
LONG Jun-Ying1MA Lan1,2HE De-Hua1,*
(1Innovative Catalysis Program,Key Laboratory of Organic Optoelectronics and Molecular Engineering of Ministry of Education,Department of Chemistry,Tsinghua University,Beijing 100084,P.R.China;2Institute of Chemical Defence,Beijing 102205,P.R.China)
Co-B catalysts supported on ZrO2(prepared by alcogel nitrogen thermo-treatment method),carbon nanotubes (CNTs),and mesoporous SBA-15 as supports were prepared by chemical reduction method and characterized by X-ray diffraction(XRD),nitrogen physisorption,transmission electron microscopy(TEM),inductively coupled plasma-atomic emission spectrometry(ICP-AES)and X-ray photoelectron spectroscopy(XPS).Hydroformylation of 1-octene was used as a probe reaction to investigate the catalytic performance and stability of the catalysts in recycles.The asprepared Co-B catalysts showed amorphous characteristics and the support structures were unchanged.The reaction results reveal that the catalytic activities of Co-B/CNTs,Co-B/ZrO2and Co-B/SBA-15 in the hydroformylation of 1-octene were comparatively high.However,the catalytic activities decreased upon recycling.Nevertheless,the Co-B/ CNTs and Co-B/SBA-15 were relatively stable compared with Co-B/ZrO2.
Amorphous Co-B catalyst;Support;Hydroformylation;Catalytic property
O643
Received:April 26,2010;Revised:June 24,2010;Published on Web:September 10,2010.
*Corresponding author.Email:hedeh@mail.tsinghua.edu.cn;Tel/Fax:+86-10-62773346.
The project was supported by the National Natural Science Foundation of China(20921001).
國家自然科學基金(20921001)資助項目
?Editorial office of Acta Physico-Chimica Sinica
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
