
酯加氫反應中影響羧基活化的因素
2010-11-30 10:50:08鄭小娟周婭芬付海燕李賢均李瑞祥
物理化學學報 2010年10期
關鍵詞:催化劑
鄭小娟 周婭芬 付海燕 陳 華 李賢均 李瑞祥
(四川大學化學學院,教育部綠色化學與技術重點實驗室,成都 610064)
酯加氫反應中影響羧基活化的因素
鄭小娟 周婭芬 付海燕 陳 華 李賢均 李瑞祥*
(四川大學化學學院,教育部綠色化學與技術重點實驗室,成都 610064)
通過浸漬法制備4%Ru-9%La/γ-Al2O3催化劑,采用X射線衍射(XRD),X射線光電子能譜(XPS)和透射電子顯微鏡(TEM)對其結構進行表征.將該催化劑用于丙酸甲酯的加氫反應,分別考察了溶劑、無機鹽添加劑、底物空間因素及電子因素對酯加氫反應的影響.發現以水為溶劑以及添加Co(NO3)2對丙酸甲酯加氫都顯示出明顯的促進作用,底物的轉化率及丙醇的選擇性隨之增加.這是由于水及適量Co2+的引入能極化底物分子的C=O鍵,有利于活化氫對羧基碳原子的進攻,改善催化劑的性能.此外,底物分子中吸電子基團能提高羧基碳原子的正電性,也有利于加氫反應的進行;而增大底物分子空間位阻,不利于底物在催化劑上的吸附,催化反應速率下降.
催化加氫;丙酸甲酯;活化;釕; 鑭
醇是化學和醫藥工業中重要的中間體,可以由含羰基的化合物如醛、酮、羧酸及其酯或酐加氫得到.然而由于酯基中C=O雙鍵有較大的空間位阻和弱的極性使得羧酸酯還原成醇十分困難[1].工業上通常用銅鋅、銅鉻等復合氧化物催化羧酸及其酯加氫[2-3],加氫需要在高溫(250-300℃)和高壓(25-35 MPa)下進行.后來,人們對釕、銠等貴金屬作為主要活性組分的催化劑用于酯加氫進行了研究.但單金屬催化劑對羧酸及酯的加氫活性和選擇性往往不高[4],為此,人們通過引入一種或多種金屬來調變單金屬催化劑的性能,對酯加氫取得了較高的活性和醇選擇性.例如用Rh-Sn/SiO2,Ni-Sn/SiO2和Ru-Sn/ SiO2雙金屬催化劑成功對乙酸酯進行了氣相加氫制備乙醇[5].雙金屬催化劑Ru-Sn/SiO2[5]、Ru-Sn/TiO2[6-7]、Ru-Sn-B/Al2O3[8]、Co-Sn/Al2O3[9]、Ru-Sn/Al2O3[10-11]、Rh-Sn/Al2O3[12]被廣泛地用于直鏈羧酸及酯的加氫,取得了不錯的效果.文獻報道[11,13]負載Ru-Sn催化劑催化酯加氫反應中,SnOx物種(Sn2+或Sn4+)作為Lewis酸與羧基氧結合極化了C=O鍵,從而改善了酯加氫的性能.本課題組[14]用Ru-Pt/AlO(OH)對丙酸甲酯加氫發現,溶劑H2O和載體AlO(OH)的表面羥基與底物的羧基氧能形成氫鍵,同樣可以極化C=O鍵,并且加速了反應的進行.本文在此基礎上,以丙酸甲酯為模型化合物,進一步研究在溶液中添加無機鹽,以及底物空間和電子因素對羧基活化的影響.
1 實驗部分
1.1 試 劑
γ-Al2O3(AR,100-160目,山西日化所,500℃焙燒4 h);RuCl3·nH2O(AR,昆明貴金屬研究所);LaCl3· 7H2O、SnCl4、SnCl2、ZnSO4、Co(NO3)2、Ca(NO3)2、FeSO4、FeCl3均為分析純;乙醇、正丙醇、異丙醇、正丁醇(AR,成都長聯化工試劑有限公司);丙酸甲酯、丙酸乙酯、丙酸、乙酸乙酯、三氟乙醇、三氟乙酸乙酯等均為分析純.丙酸正丙酯、丙酸異丙酯、丙酸正丁酯、丙酸三氟乙酯自制;氫氣純度大于99.99%(四川天一科技股份有限公司).
1.2 催化劑的制備及表征
4%Ru-9%La/γ-Al2O3制備.將一定量的LaCl3· 7H2O水溶液加入到γ-Al2O3中,室溫攪拌過夜后旋干,120℃干燥24 h,350℃煅燒3 h,得催化劑前驅體La2O3/γ-Al2O3.將一定量的RuCl3溶液加入到懸浮了La2O3/γ-Al2O3的水溶液中,室溫下攪拌過夜后旋干,轉入高壓釜中以水為溶劑,在180℃和5.0 MPa條件下,加氫還原3 h.固體物質經過濾,去離子水洗滌至無氯離子,60℃真空干燥8 h,即得催化劑.洗滌后的濾液收集用于測定金屬擔載量,經電感耦合等離子體光譜(ICP)測得金屬擔載的Ru的質量分數為4%,La2O3/γ-Al2O3質量比為9%.
催化劑的X射線光電子能譜(XPS)在英國Kratos公司的XSAM 800型X射線光電子能譜儀上測得,激發源為Al Kα(1215 eV),電流15 mA,電壓12 kV,用C 1s結合能284.8 eV定標.X射線衍射(XRD)采用日本理學D/max-rA旋轉陽極X射線衍射儀,Cu Kα射線,λ=0.154 nm,管電壓50 kV,管電流40 mA,掃描區間為20°-110°.催化劑的形貌與金屬粒子大小通過日本JEOL公司的JEM-1200EX型透射電子顯微鏡測定,加速電壓為100 kV.金屬擔載量用ICP(美國熱電元素公司IRIS Advantage型光譜儀)測定.
1.3 催化加氫反應
底物的催化加氫反應在60 mL帶磁力攪拌裝置的不銹鋼高壓釜中進行.將一定量催化劑、反應底物和溶劑加入高壓釜中,閉釜后用高純氫置換反應釜5次,充氫氣至所需壓力,升溫至設定溫度開始攪拌并計時(攪拌速度為1500 r·min-1).反應結束后,取出樣品,過濾除去催化劑,加氫產物在安捷倫GC-6890氣相色譜儀上分析,SE-30石英毛細管柱(30 m×0.25 mm×0.25 μm,美國Supelco公司).采用峰面積歸一化法計算含量.
2 結果與討論
2.1 催化劑的表征
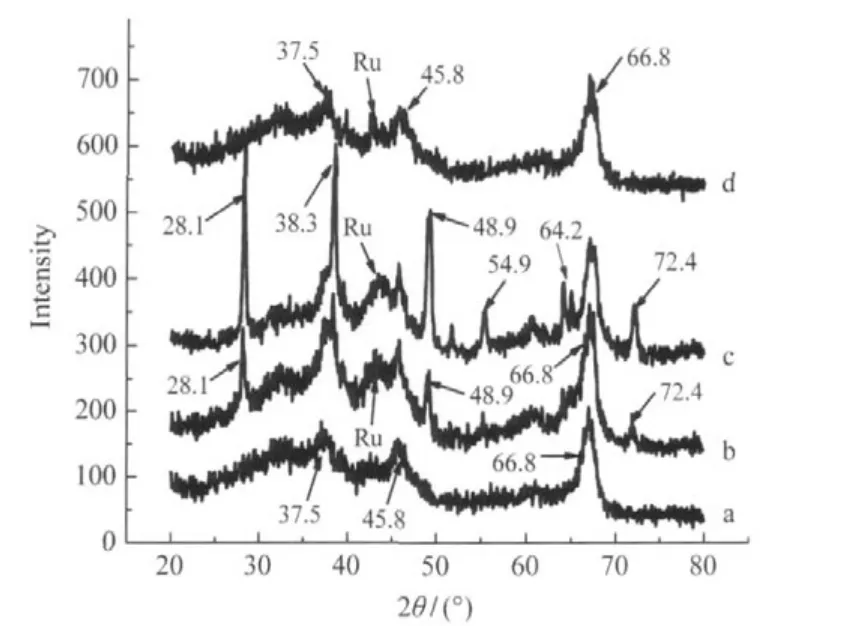
圖1 4%Ru-9%La/γ-Al2O3催化劑的XRD圖Fig.1 XRD patterns of 4%Ru-9%La/γ-Al2O3catalyst (a)not reduced,(b)reduced with H2in water for 3 h,(c)afterhydrogenation in water for 8 h;(d)reduced with H2in ethanol for 3 h; (b-d)temperature:180℃,H2pressure:5.0 MPa
催化劑4%Ru-9%La/γ-Al2O3樣品a-d經不同處理:未經還原(樣品a);以水做溶劑、180℃、5.0 MPa H2下還原3 h(樣品b);還原、并用水做溶劑、180℃、5.0 MPa H2下反應8 h(樣品c)以及以乙醇做溶劑、180℃、5.0 MPa H2下還原3 h(樣品d)后的XRD譜見圖1.圖1a中2θ為37.5°、45.8°、66.8°的彌散峰屬于γ-Al2O3的衍射峰(JCPDS No.29-63),可能由于金屬擔載量低分散好的緣故,均沒有觀察到Ru、RuO、Ru2O的衍射峰[15].圖1b和圖1c中2θ為28.1°、38.3°、48.9°、54.9°、64.2°及72.4°的峰為薄水鋁石的衍射峰(γ-AlO(OH),JCPDS No.21-1307).表明催化劑在水熱條件下還原以及水作溶劑條件下反應后,γ-Al2O3轉變成為AlO(OH)[16].而且反應8 h后的催化劑,這些峰變得更強更尖銳,說明AlO(OH)在催化反應過程中進一步生長變大.圖1d中只發現2θ為37.5°、45.8°、66.8°的γ-Al2O3衍射峰,并沒發現薄水鋁石的衍射峰,說明催化劑在乙醇中還原時,γ-Al2O3沒有轉變成γ-AlO(OH).而且還原和使用后催化劑的XRD圖中(圖1(b,c,d)),在2θ=43°處出現一個弱而彌散的Ru0衍射峰(JCPDS No.6-0663),表明金屬顆粒在加氫過程中發生了團聚.
圖2為催化劑未經還原(圖2a),以水為溶劑、在180℃、5.0 MPa H2條件下還原3 h(圖2b),及以乙醇為溶劑、在180℃、5.0 MPa H2條件下還原3 h(圖2c)后的TEM圖.從圖2b中可以看到大的薄片狀薄水鋁石載體 ,同時也有少量未完全轉化的γ-Al2O3,而圖2(a,c)中載體均為纖維狀的γ-Al2O3,這與XRD譜圖結果一致.另外,從幾個圖上都可看到,金屬粒子約為2-5 nm,且均勻分布于載體上.
圖3是該催化劑反應后(水做溶劑,180℃、5.0 MPa H2,反應時添加Co(NO3)2)的XPS圖譜,從圖3a上可見Ru 3d5/2的結合能位于280.13 eV,與Ru0的結合能十分接近,證明催化劑中的Ru以零價態形式存在[18].而La 3d5/2(圖3b)的電子結合能834.89 eV,與文獻中La2O3(834.92 eV,La3+)的值[19]幾乎一樣,說明反應后鑭的氧化態沒有發生變化.Co的電子結合能為781.6 eV(圖3c),表明Co在反應后氧化態也沒有發生變化[20].
2.2 溶劑對酯加氫反應的影響

圖2 不同催化劑的TEM圖Fig.2 TEM images of different catalysts (a)not reduced;(b)reduced with H2in water for 3 h;(c)reduced with H2in ethanol for 3 h (b,c)temperature:180℃,H2pressure:5.0 MPa

圖3 4%Ru-9%La/γ-Al2O3催化劑在水中反應8 h后的XPS圖譜Fig.3 XPS spectra of the 4%Ru-9%La/γ-Al2O3catalyst after hydrogenation in water for 8 h temperature:180℃,H2pressure:5.0 MPa
溶劑對丙酸甲酯加氫反應的影響的結果列于表1.當催化劑在水中以H2還原,催化反應以水作溶劑時(entry 1),反應表現出高的活性和醇選擇性.而以乙醇或乙醇-水混合物作溶劑時(entry 2和entry 3),不僅轉化率比水作溶劑時低,而且生成較多的酯交換產物.說明水能抑制酯交換反應的發生[21].由entry 1和entry 4結果可以看出,以水作反應溶劑時,在水中還原的催化劑又比在乙醇中還原的催化劑活性更高.根據XRD、TEM表征結果,乙醇中還原的催化劑,載體在還原過程中結構沒有發生變化,仍為γ-Al2O3;而在水中還原的催化劑,γ-Al2O3轉變成了薄水鋁石.我們以前研究Ru-Pt/AlO(OH)對丙酸甲酯加氫反應發現,載體豐富的表面羥基和溶劑水的協同作用能促進反應活性和選擇性的提高.一方面, AlO(OH)的表面羥基與底物的羧基氧形成氫鍵,極化了C=O鍵,使Ru上活化的氫對C=O鍵上的C原子進攻更加容易[14];另一方面,水分子中堿性氧原子易于與配位H2結合,促進其異裂得到Ru—H和質子化的水[22-23],然后Ru上H-進攻羧基上缺電子的C原子形成中間產物醛[13],醛迅速加氫得到產物醇.而本文制備的催化劑在水熱條件下轉化為Ru-La/AlO(OH),表現出相似的性質.
2.3 無機鹽添加劑對酯加氫反應的影響
通過溶劑的選擇研究發現,底物與溶劑間形成氫鍵可以活化羧基,使加氫反應更易發生[14].而金屬離子對—NO2的活化作用也曾有文獻報道 ,認為金屬離子作為路易斯酸能與—NO2中作為路易斯堿的氧原子作用,從而活化—NO2中的N=O鍵,在加氫條件下實現了—NO2快速還原為—NH2.在文獻報道的Ru-Sn雙金屬催化酯加氫機理中[11,13],作者同樣認為SnOx物種(Sn2+或Sn4+)作為路易斯酸與酯中羧基氧結合極化了C=O鍵,從而利于活化氫對C=O鍵上碳原子的進攻.因此,我們為了更清楚地理解羧基活化的原理,將金屬離子直接添加到反應溶液中,考察其對酯加氫的影響.
表2列出了各種無機鹽添加劑對丙酸甲酯加氫反應的影響.數據顯示,添加適量的Co2+能明顯提高底物的轉化率,醇的選擇性也保持不變.當添加的Co(NO3)2溶液使得Co/Ru摩爾比為4時,丙酸甲酯的轉化率得到了最大程度的提高,從82.2%上升到了96.8%.繼續增大Co/Ru比,底物的轉化率和醇選擇性都降低.然而其它的添加劑對催化活性沒有明顯改善,有些反而還使活性降低.
為了進一步探討Co2+在反應中是如何對反應起促進作用時,我們進一步考察了Co2+的添加方式對反應的影響,結果見表3所示.方式I是將RuCl3和Co(NO3)2溶液共浸漬到催化劑前體La/γ-Al2O3上,然后300℃煅燒4 h制得;方式Ia是與I同樣方式負載但不煅燒.方式II是將制成的Ru-La/γ-Al2O3催化劑再浸漬Co(NO3)2溶液,最后300℃煅燒4 h制得;IIa是與II同樣方式負載但不煅燒.方式III是用已制備好的Ru-La/γ-Al2O3作催化劑,將Co(NO3)2直接加入反應的溶液中.
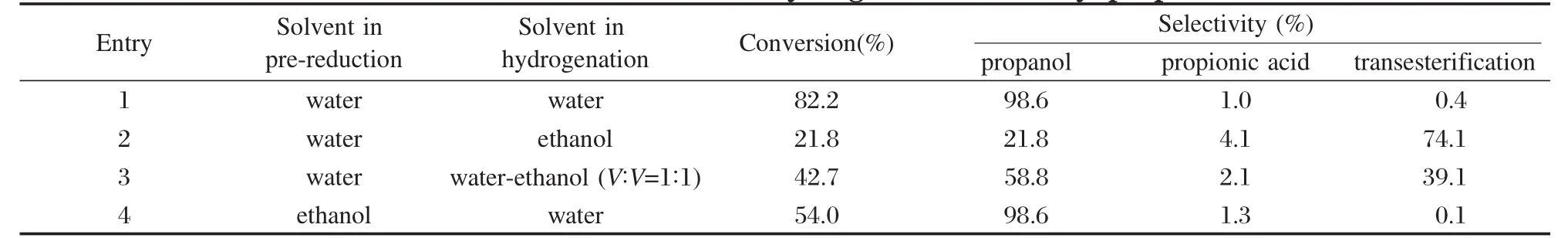
表1 溶劑對丙酸甲酯加氫反應的影響Table 1 Effect of solvent on the hydrogenation of methyl propionate
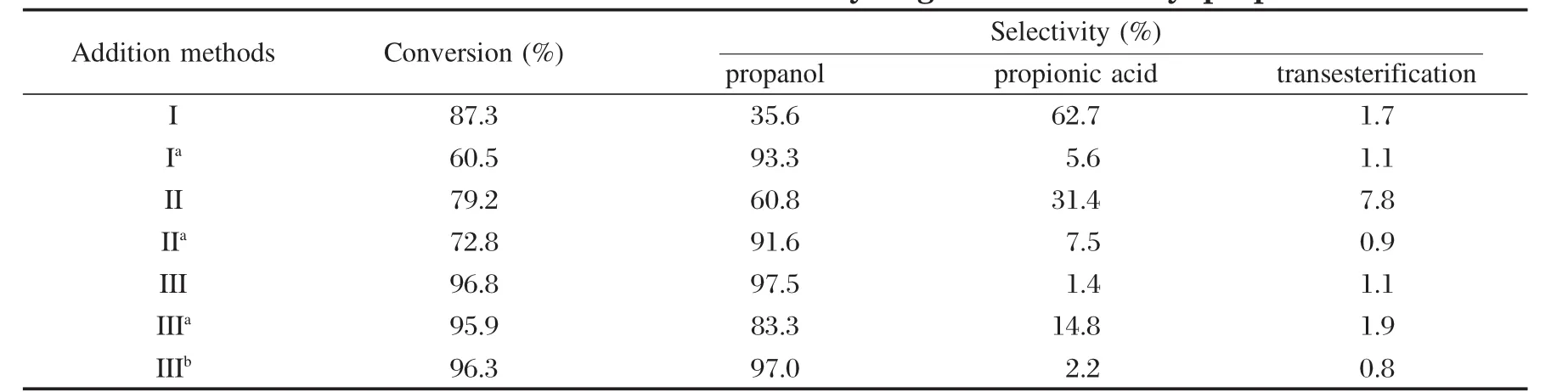
表3 Co不同添加方式對丙酸甲酯加氫的影響Table 3 Effect of Co2+addition method on hydrogenation of methyl propionate
表3數據顯示,Co2+添加到溶液中的效果最好,而方法I和II都使催化活性降低.比較I和Ia、II和IIa發現,通過煅燒引起了催化劑活性明顯降低,選擇性也大幅下降.不經過煅燒,催化劑的活性下降,選擇性下降不明顯.由此可見,將Co2+全部吸附到催化劑上對酯加氫反應并沒有促進作用.這些結果表明,經煅燒后Co(NO3)2將轉化為氧化鈷;將Co(NO3)2吸附在催化劑上不煅燒,鈷將仍以離子態存在于催化劑表面.雖然氧化鈷存在時,底物轉化率比離子態的Co2+存在的催化劑高,但底物主要轉化成了丙酸,生成目標產物的量很少.大量離子態的Co2+存在于催化劑表面時,催化劑的活性和選擇性也沒有改善.
由于Co2+被添加到水溶液中表現出最好的性能,為了研究清楚Co2+是在溶液中還是吸附在催化劑表面起作用,我們將三種方式反應后的溶液分別進行ICP分析,發現方式I和II反應后的濾液中只有極少量的Co2+,99.8%的Co2+都保持在催化劑表面;IIa反應后溶液中鈷含量經ICP分析為2.0%,可見在催化劑表面吸附的Co2+對反應沒有促進作用.而III反應后的溶液中存在17%的Co2+,有83%的Co2+吸附到了催化劑表面.我們又將III反應后的催化劑分離并再一次反應:一種是直接使用III反應后的催化劑(IIIa),不補加Co(NO3)2;另一種(IIIb)是用III反應后的催化劑,再補加17%的Co(NO3)2(與III反應后溶液中的Co2+量相同).從表3結果可以看出,在反應液中補加Co(NO3)2比不補加對生成丙醇的選擇性要高.IIIa反應的溶液中有5.7%的Co2+存在,這是催化劑上吸附的Co2+再次進入溶液的緣故. IIIb反應后的濾液中存在10.2%的Co2+,比IIIa反應后溶液中的Co2+多了4.5%.從上述結果看出,溶液中Co2+量多,轉化率和選擇性則明顯提高.上述結果說明了Co2+對反應的促進作用是發生在溶液中的.
另外,III反應后催化劑的XPS測試(圖3)表明催化劑上沒有金屬態的Co,這說明在反應中是Co2+起促進作用.我們認為添加的Co2+也是作為路易斯酸.以水為溶劑時,丙酸甲酯在水中有一定的溶解性,溶于水中的Co2+和溶于水中的酯的羧基能發生配位作用,導致電負性大的氧原子上電荷密度下降,從而極化了C=O,促進了反應的進行.
2.4 電子因素、空間因素對酯加氫反應的影響
為了進一步證明羧基碳原子上正電荷密度增加有利于羧基的活化,我們以4%Ru-9%La/γ-Al2O3為催化劑,考察了底物電子因素、空間因素對反應的影響,結果列于表4.從表4中可以看出,隨著底物分子中基團的增大(entry 1到entry 5),發現無論是羧酸鏈的增大,還是酯基的增大所引起的空間位阻都不利于底物在催化劑表面的吸附,因此加氫活性降低.再分別將entry 2和entry 6、entry 7和entry 8進行比較發現,將底物分子(entry 2和entry 7)中的—CH3變成—CF3(entry 6和entry 8)時,底物的轉化率明顯增加.由于—CF3基作為吸電子基團,—CF3的存在可以增加底物分子中羧基碳原子上的正電荷密度,同樣起到了極化了C=O鍵的目的,因而有利于活化的氫進攻C=O鍵上的碳原子,使反應活性增強.
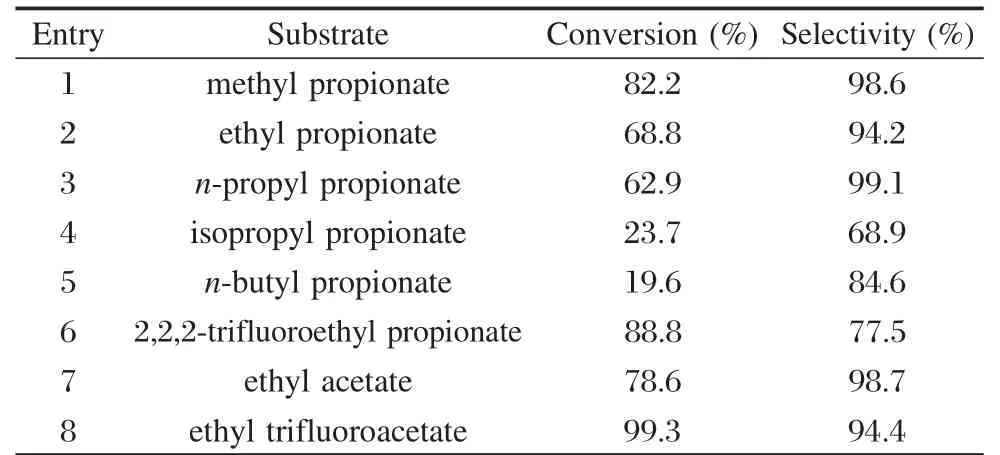
表4 催化劑對不同底物的加氫性能Table 4 Hydrogenation performance of the catalyst for different substrates
3 結 論
制備了Ru-La/γ-Al2O3催化劑,并用于丙酸甲酯的加氫反應,分別考察了溶劑、無機鹽添加劑、底物的電子因素等對酯加氫的影響.發現以水為溶劑時,反應條件下γ-Al2O3轉化成AlO(OH)、AlO(OH)的表面羥基與底物羧基氧形成的氫鍵可以引起羧基活化.而在反應溶液中添加適量的Co(NO3)2,鈷作為路易斯酸能與酯的羧基氧發生配位作用,導致羧基氧原子上電荷密度下降,從而極化了C=O鍵.此外,底物分子中吸電子基團導致羧基碳原子上正電荷密度增加,同樣有利于羧基的活化.因此羧基C=O鍵的極化或碳原子正電荷的增加是引起酯加氫活性改善的重要因素.
1 McAlees,A.J.J.Chem.Soc.C,1969:2425
2 Rieke,R.D.;Thakur,D.S.;Roberts,B.D.;White,G.T.J.Am.Oil Chem.Soc.,1997,74(4):333
3 Zhu,J.F.Preparation and characterization of middle-pressure hydrogenation catalyst for fatty acid methyl esters[D].Qingdao: China University of Petroleum(East China),2007 [朱建鋒.脂肪酸甲酯中壓加氫催化劑制備與表征[D].青島:中國石油大學(華東),2007]
4 Luo,G.;Yan,S.R.;Qiao,M.H.;Zhang,J.H.;Fan,K.N.Appl. Catal.A:Gen.,2004,275:95
5 Bournonville,J.P.;Mabilon,G.;Candy,J.P.;Basset,J.M.J.Mol. Catal.,1991,67:283
6 Corradini,S.A.S.;Lenzi,G.G.;Lenzi,M.K.;Soares,C.M.F.; Santos,O.A.A.Journal of Non-Crystalline Solids,2008,354: 4865
7 Silva,A.M.;Morales,M.A.;Baggio-Saitovitch,E.M.;Jord?o,E.; Fraga,M.A.Appl.Catal.A-Gen.,2009,353:101
8 Piccirilli,A.;Pouilloux,Y.;Pronier,S.;Barrault,J.Bull.Soc. Chim.France,1995,132:1109
9 Pouilloux,Y.;Autin,F.;Barrault,J.Catal.Today,2000,63:87
10 Silva,A.M.;Santos,O.A.A.;Morales,M.A.;Baggio-Saitovitch, E.M.;Jord?o,E.;Fraga,M.A.J.Mol.Catal.A,2006,253:62
11 Santos,S.M.;Silva,A.M.;Jord?o,E.;Fraga,M.A.Catal.Today, 2005,107-108:250
12 Miyake,T.;Makino,T.;Taniguchi,S.I.;Watanuki,H.;Niki,T.; Shimizu,S.;Kojima,Y.;Sano,M.Appl.Catal.A-Gen.,2009, 364:108
13 Pouilloux,Y.;Autin,F.;Guimon,C.;Barrault,J.J.Catal.,1998, 176:215
14 Zhou,Y.F.;Fu,H.Y.;Zheng,X.J.;Li,R.X.;Chen,H.;Li,X.J. Catal.Commun.,2009,11:137
15 Hu,S.C.;Chen,Y.W.J.Chem.Technol.Biotechnol.,2001,76: 954
16 Okada,K.;Nagashima,T.;Kameshima,Y.;Yasumori,A.; Tsukada,T.J.Colloid Interface Sci.,2002,253:308
17 Chen,X.Y.;Lee,S.W.Chem.Phys.Lett.,2007,438:279
18 Wang,J.Q.;Wang,Y.Z.;Xie,S.H.;Qiao,M.H.;Li,H.X.;Fan, K.N.Appl.Catal.A,2004,272:29
19 Fleisch,T.H.;Hicks,R.F.;Bell,A.T.J.Catal.,1984,87:398
20 Struijk,J.;Scholten,J.J.F.Appl.Catal.A,1992,82:277
21 Wehner,P.S.;Gustafson,B.L.J.Catal.,1992,135:420
22 Clapham,S.E.;Hadzovic,A.;Morris,R.H.Coord.Chem.Rev., 2004,248:2201
23 Nomura,K.;Ogura,H.;Imanishi,Y.J.Mol.Catal.A,2001,166: 345
24 Zuo,B.J.;Wang,Y.;Wang,Q.L.;Zhang,J.L.;Wu,N.Z.;Peng, L.D.J.Catal.,2004,222:493
25 Xu,Q.;Liu,X.M.;Chen,J.R.;Li,R.X.;Li,X.J.J.Mol.Catal.AChem.,2006,260:299
April 29,2010;Revised:June 3,2010;Published on Web:July 15,2010.
Activation Factors for the Carboxyl Group in the Hydrogenation of Carboxylic Esters
ZHENG Xiao-Juan ZHOU Ya-Fen FU Hai-Yan CHEN Hua LI Xian-Jun LI Rui-Xiang*
(Key Laboratory of Green Chemistry and Technology,Ministry of Education,College of Chemistry,Sichuan University, Chengdu 610064,P.R.China)
We prepared a 4%Ru-9%La/γ-Al2O3catalyst by impregnation method and characterized it using X-ray diffraction(XRD),X-ray photoelectron spectroscopy(XPS)and transmission electron microscopy(TEM).The catalyst was used for the hydrogenation of methyl propionate.The effects of solvent,inorganic salt additive,steric as well as electronic factors of the substrate on the hydrogenation of a carboxylic ester were investigated.We found that both water and the Co(NO3)2additive obviously improved the hydrogenation of methyl propionate,the conversion of the substrate and the selectivity for propanol.The promotional effects of water and Co2+are attributed to polarization of the C=O bond in the carboxyl group of the substrate molecule by the formation of a hydrogen bond between water and the carboxylic group and the coordination of Co2+to the carboxylic group.This is favorable for an attack on the carbon atom of the carboxyl group by the activated hydrogen.Similarly,the electron-withdrawing group in the substrate molecule also caused the high positive charge of carbon in the carboxyl group.The highly positive charged carbon is beneficial for the hydrogenation reaction.In addition,an increase in the steric hindrance of the substrate molecules was not favorable for the adsorption of the substrate on the catalyst and,therefore,the reaction rate decreased.
Catalytic hydrogenation;Methyl propionate;Activation;Ruthenium;Lanthanum
O643
*Corresponding author.Email:sculiruixiang@163.com;Tel:+86-28-85412904.
The project was supported by the National Natural Science Foundation of China(21072138).
國家自然科學基金(21072138)資助項目
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
