焦化蠟油兩段提升管催化裂解多產丙烯與焦化汽油改質研究
2011-01-13 08:29:54孫金鵬山紅紅李春義陳小博
石油煉制與化工 2011年11期
孫金鵬,山紅紅,李春義,陳小博
(中國石油大學 (華東)化學化工學院重質油國家重點實驗室,青島266555)
1 前 言
隨著常規原油資源的枯竭,重油資源越來越受到人們的關注。長期以來,對于劣質重油加工的一個重要手段就是直接進入延遲焦化裝置[1-2]。根據粗略統計,截止到2009年底,中國延遲焦化加工能力已經達到99.05Mt/a,在建裝置規模為9.40Mt/a。由于焦化裝置產能的急劇增加,焦化產物如焦化蠟油(CGO)等劣質原料的加工也成為眾多研究者重點關注的研究方向,延遲焦化-催化裂化組合工藝的開發一直是研究的熱點[3-6]。然而CGO作為催化裂化原料卻存在著多方面的問題,例如CGO中氮化物含量,尤其是堿氮化合物含量非常高,而催化裂化催化劑是酸性催化劑,堿氮吸附到催化劑酸性位上,對于催化裂化催化劑有直接的毒害作用,直接影響原料的轉化率和汽油、柴油收率[7-8];CGO中稠環芳烴等難裂解組分含量較高,因此,CGO催化裂化回煉量較大,為提高反應深度,不得不提高反應溫度,使得焦炭及干氣產率增加。中國石油大學重質油國家重點實驗室自主開發了兩段提升管催化裂解多產丙烯技術(TMP)[9-12],通過采取組合進料及適宜的操作條件,以及自主開發的新型多產丙烯催化劑,較好地解決了普通催化裂化工藝在多產液化氣時干氣產率較高的難題。本研究主要考察CGO在TMP工藝條件下的催化裂解性能,以及CGO催化裂解與焦化汽油(CN)改質的耦合反應性能。
2 實 驗
2.1 原 料
試驗所用原料為恒源石化提供的CGO和CN。CGO的性質見表1。CN的性質見表2。從表1可以看出:CGO的H元素質量分數為11.41%,但飽和分含量較低,而芳香分、膠質及瀝青質的難裂化組分含量較高;CGO的N質量分數為0.45%,而堿氮質量分數高達1 435μg/g,表明CGO進行催化裂化的困難程度很高[7]。從表2可以看出,CN的烷烴含量較高,芳烴質量分數僅為7.98%,RON為61.9,且硫含量較高,很難將其作為汽油調合組分。
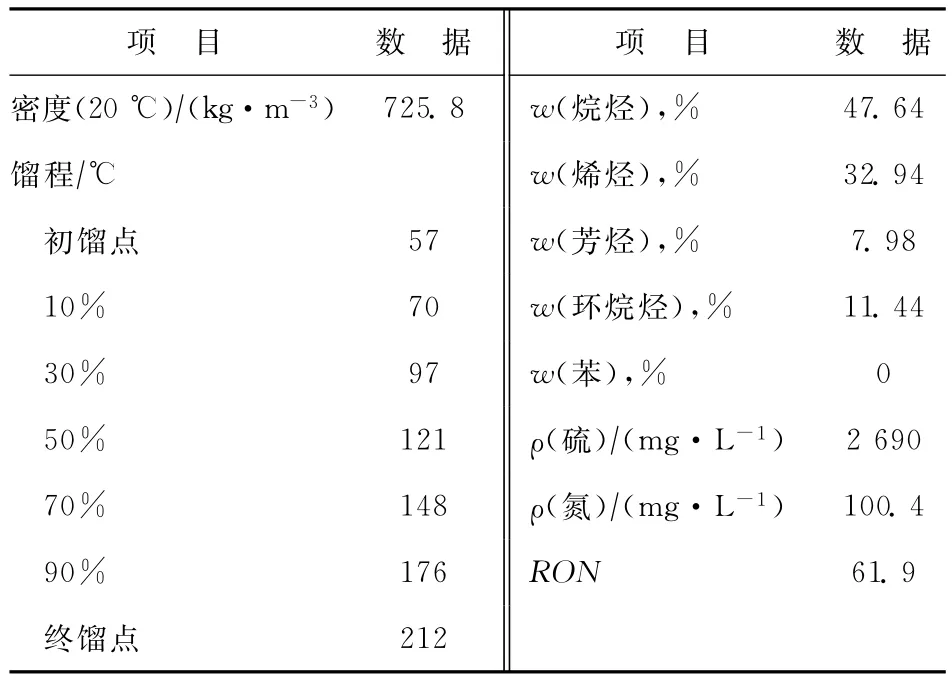
表2 CN的基本性質
2.2 催化劑
試驗所用催化劑為兩種多產低碳烯烴的催化裂解催化劑A、B按照一定比例混合而成,其中催化劑A為本課題自主研發的多產低碳烯烴催化劑(已工業化),催化劑B為中國石化石油化工科學研究院開發的多產低碳烯烴催化劑,兩種催化劑的物理性質見表3。
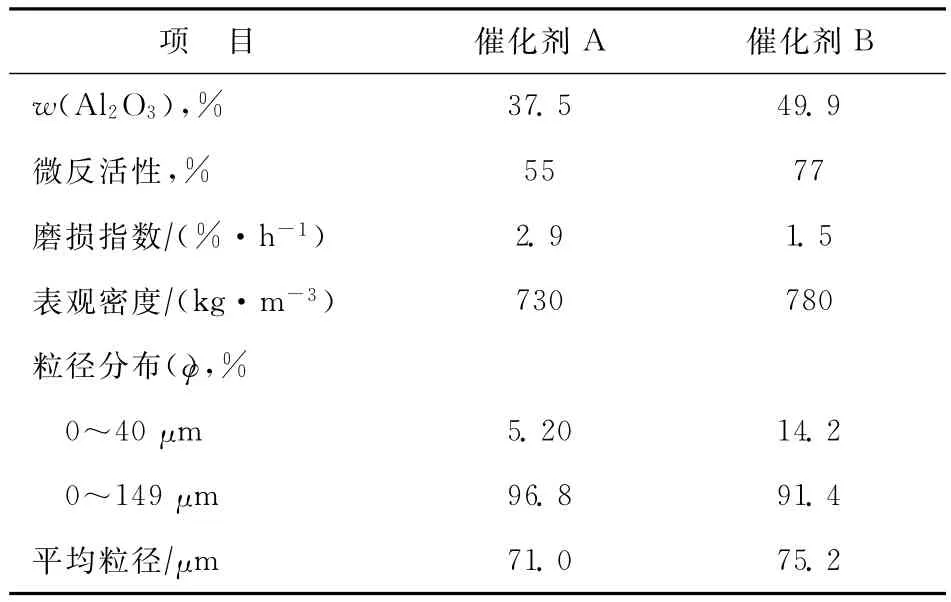
表3 催化劑的物理性質
2.3 試驗裝置
試驗在ZDT-1中型提升管催化裂化試驗裝置上進行,中試裝置示意見圖1。該裝置可以模擬工業裝置,實現催化劑的連續反應-再生循環;利用該裝置可進行工業催化裂化裝置的工藝條件模擬試驗、各種原料裂化性能及其產品性質的考察、催化劑的評價以及反應再生動力學等方面的試驗研究。氣體分析與液體分析均在Varian CP3800型氣相色譜儀上完成。

圖1 催化裂化組合進料中試裝置示意
3 結果與討論
3.1 CGO兩段提升管催化裂解
兩段提升管催化裂解反應是指第一段反應后的液體產品經抽出沸點小于350℃的汽油、柴油餾分后的重油再進入第二段反應。CGO兩段催化裂解產物分布及兩段反應數據綜合計算結果見表4。從表4可以看出:在反應溫度520℃、停留時間1.2s,劑油質量比11的條件下,CGO轉化率達到73.40%,液化氣產率達到31.62%;丙烯產率為15.78%,約占液化氣的50%左右,干氣產率僅為3.19%。由于第二段難裂化組分較多,因此在第二段反應時稍微提高操作苛刻度。由兩段反應數據綜合計算結果可知:兩段提升管裂解后轉化率為87.80%,較一段反應提高14.40百分點,說明一段重油轉化仍有較大潛力可挖,可能是原料中輕膠質起到了關鍵作用[10],因此避免一段轉化率過高,可以有效地避免一段重油在第二段較難轉化,以至于使第二段反應干氣及焦炭產率過高的情況;兩段提升管裂解后,丙烯收率為18.12%,較一段反應提高了2.34百分點,同時還產出了14.21%的丁烯,這對于丁烯回煉進一步增產丙烯是有利的。從表4還可以看出,兩段提升管裂解后,液化氣收率達到35.90%,丙烯和丁烯兩種產物對液化氣的選擇性超過了90%,干氣產率僅為4.27%,這與實驗所使用的高選擇性多產丙烯催化劑有密切關系。ARGG技術[13]的丙烯產率一般為7%~10%,而DCC技術采用品質很好的石蠟基原料時丙烯收率最高可達23%,但此時干氣收率在8%以上[14]。在本研究中,除了液化氣產率較高外,還產出了20.35%的汽油。

表4 CGO催化裂解反應產物分布
兩段提升管裂解反應后的汽油族組成見表5。從表5可以看出,汽油中烯烴質量分數為49.09%,含量較高,芳烴質量分數為23.04%。而PONA數據表明,汽油中C=5質量分數達到34.37%,C=6質量分數達到9.30%。因此通過輕汽油回煉可以在增產丙烯的同時,降低汽油中的烯烴含量,也會相應地提高汽油中的芳烴含量,從而提高汽油品質。

表5 兩段提升管裂解汽油族組成 w,%
3.2 CGO催化裂解與CN改質耦合
CN是延遲焦化的另一種主要產物,在熱裂解條件下,烴類主要發生分解與縮合兩類反應,很少發生異構化、氫轉移及芳構化反應,因而CN中正構烷烴含量較高,且有較多的不飽和烴保留下來,而異構烷烴和芳烴含量相對較低,所以CN的辛烷值低,且安定性差。
一般CN可以通過加氫精制或者芳構化進行改質[15-16]。近年來,CN通過提升管進行催化裂化改質的研究越來越多[17-20]。本研究采用CGO催化裂解與CN改質耦合的方法,考察CGO裂解和CN改質效果。
3.2.1 CN單獨改質 不同苛刻度下CN單獨改質的產物分布見表6。從表6可以看出,隨著操作苛刻度的增加,干氣產率增加,而汽油產率下降,同時焦炭產率上升。這是由于隨著苛刻度的增加,尤其是停留時間的延長,造成熱裂化反應加劇,相對于催化裂化反應而言,熱裂化反應增長的程度遠大于催化裂化反應,因此造成汽油收率下降,而低價物產率上升。因此在控制改質效果時,需要控制適宜的停留時間,避免汽油過度裂化,以保證汽油收率,盡量減少干氣等低價物的生成。同時保證較大的劑油比,使得汽油與催化劑接觸充分,促進CN中的烯烴發生裂化、異構化及芳構化反應,從而在降低汽油烯烴含量的同時,提高汽油中的芳烴及異構烷烴含量,達到提高汽油辛烷值的目的。
不同反應條件下CN改質前后族組成及辛烷值對比見表7。從表7可以看出,雖然經改質后CN中烯烴含量明顯下降,且芳烴含量也有一定程度的提高,但烷烴含量依然較高,異構烷烴由改質前的12.28%提高至13.85和15.85%,正構烷烴也有所增加,辛烷值(RON)僅從改質前的61.9提高至改質后的62.3和63.5,說明CN在提升管內的單獨改質對于其辛烷值的改善并不明顯。

表6 不同苛刻度下CN單獨改質產物分布
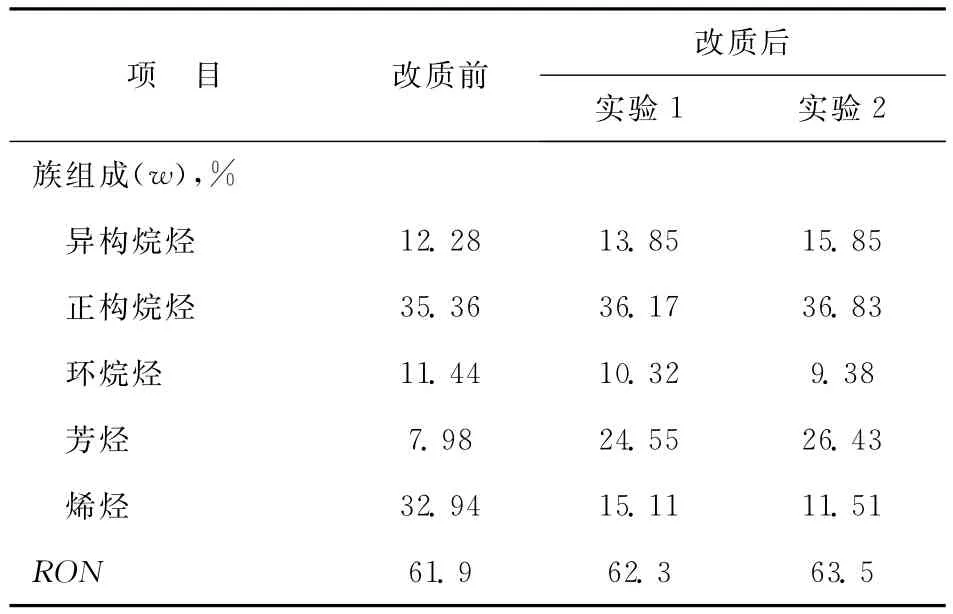
表7 不同反應條件下CN改質前后族組成及辛烷值對比
3.2.2 CGO催化裂解與CN改質的耦合 CN的平均相對分子質量比CGO小很多。根據酸性催化劑反應的正碳離子機理,大分子烴類較小分子烴類在酸性位上的吸附能力強,而且反應速率常數要大得多[21]。Torre等[22]的研究結果表明,當CN與CGO同時進料時,CN對CGO的催化裂化反應具有抑制作用。只有在較大劑油質量比(大于6)時,這種抑制作用才能避免。因此既要達到較好的CGO催化裂解效果又要達到較好的CN改質效果,CGO與CN就必須在空間與時間尺度上較為獨立,避免競爭吸附,優化產物分布,達到裂解與改質的多重耦合效應。
本研究將CN注入提升管底部,而CGO則從提升管中部進入,考察不同比例CN與CGO組合進料對CGO裂解和CN改質效果的影響,結果見表8。從表8可以看出,隨著CN比例的增加,CGO的劑油比不斷增大。由于CGO屬于劣質催化裂化原料,含有大量的稠環芳烴以及堿氮化合物[23],存在顯著的競爭吸附效應,各種組分在催化劑上吸附性能的不平均性導致了易于轉化的組分不能充分轉化。劑油比的增大實際上代表著油劑湍動混合強度的增大。油劑湍動混合強度的增大,一方面會加劇分子在催化劑活性位上的吸附-脫附頻率[24],減少雙分子反應,如氫轉移反應,從而提高丙烯選擇性,降低焦炭選擇性;另一方面提高原料油分子接觸催化劑酸性位的幾率,從而提高CGO的轉化率[25],因此大劑油比操作對于CGO轉化尤為重要。CN停留時間控制在0.5s左右,而CGO停留時間控制在1.0s左右。從產物分布來看,隨著CN進料比例的增大,干氣產率增長并不明顯,這可能一方面是由于CN停留時間很短,催化反應占主導地位,CN剛與催化劑接觸反應便已經進入到CGO反應區,而重油大分子烴類吸附能力更強,因此適時終止了汽油的反應,防止汽油的過度裂化,有效地保證了汽油收率;另一方面由于汽油在提升管底部進料,有效降低了再生劑溫度,因此在保證重油反應溫度不變時大幅度地提高了劑油比。這樣在提升管底部也形成了一個催化劑流化密度較高的汽油反應區,雖然汽油是與來自于再生器的高溫催化劑接觸,但是較高的催化劑流化密度能夠有效終止自由基反應,從而降低熱裂化程度,使得干氣產率最小化。從表8還可以看出,隨著CN進料比例的增加,液化氣收率穩定在35%左右,同時丙烯收率也穩定在16%左右,這是因為:①以ZSM-5微孔分子篩和少量USY大孔分子篩為活性組分,負載在Al2O3活性基質上,利用載體的酸性來裂解CGO中的大分子烴類,并吸附堿氮,保證了催化劑的活性;②USY分子篩保證了重油轉化率,而ZSM-5微孔分子篩孔道為0.52nm×0.54nm,可以高選擇性地多產丙烯,但是堿氮一般為2個環以上的大分子,不能進入孔道內部,從而提高了催化劑抗堿氮中毒能力,這兩點充分保證了較高的液化氣收率和較高的丙烯選擇性[26]。
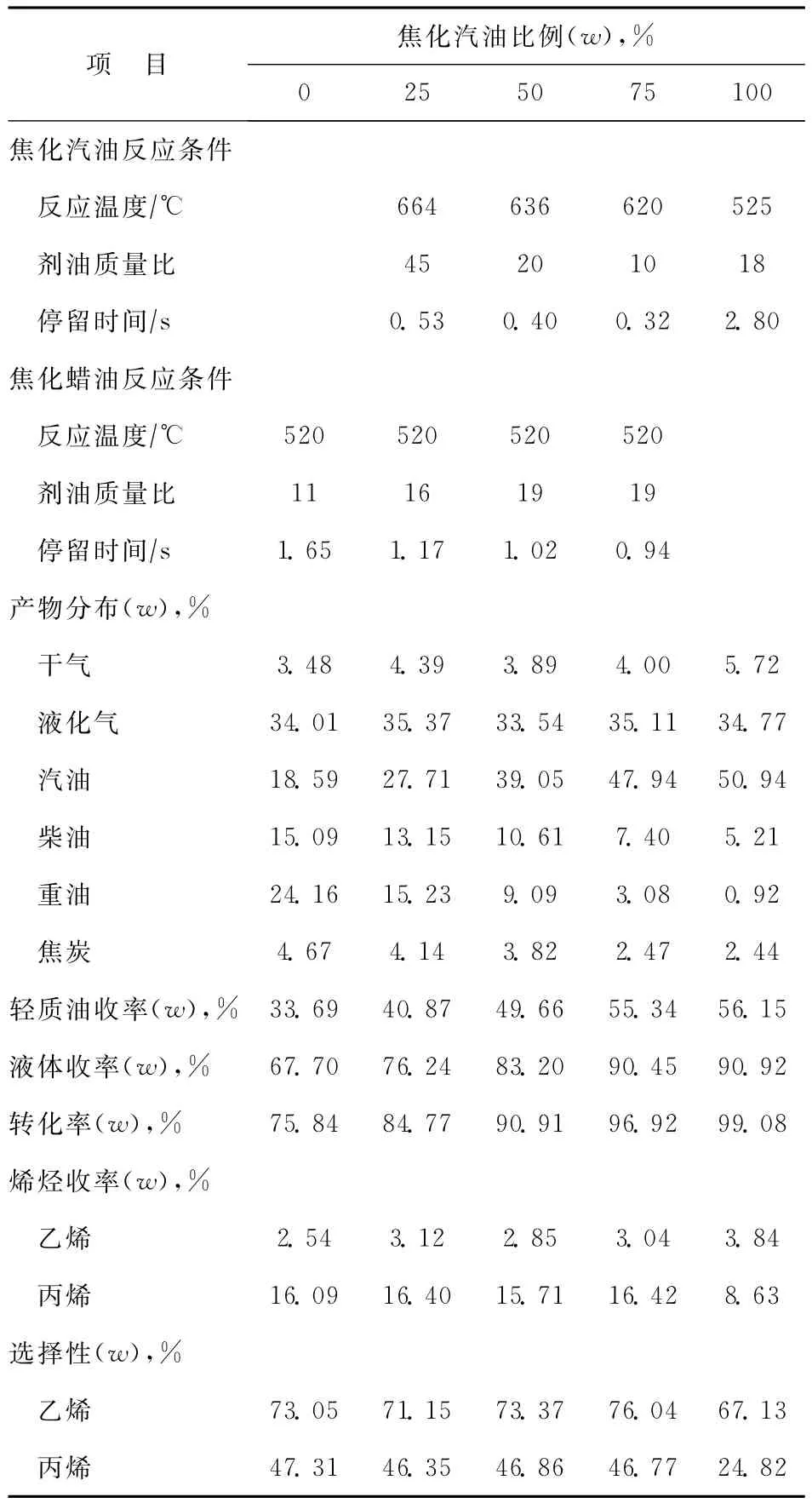
表8 CN與CGO組合進料實驗結果對比
CGO與CN組合進料汽油產品的族組成見表9。從表9可以看出,隨著CN比例的增加,汽油中烷烴含量上升,而烯烴含量迅速下降,芳烴及環烷烴含量變化不大。這是由于CN中的小分子烷烴相對較難轉化,而汽油停留時間較短,因此烷烴得以較大程度地保留。而烯烴則較為容易轉化[27]。但是單獨改質后產品汽油族組成與原料族組成的差別較大,主要是由于CN單獨改質時停留時間較長,因此部分環烷烴及烯烴發生氫轉移反應生成芳烴,而部分烯烴發生裂化反應,因此與原料族組成相比較,改質后產品汽油芳烴含量上升,烯烴含量下降明顯。

表9 CGO與CN組合進料汽油產品的族組成
CGO與CN組合進料汽油產品的研究法辛烷值見表10。從表10可以看出,隨著CN比例的增加,改質汽油的辛烷值不斷下降。但是,當CN比例為25%時,其研究法辛烷值可達到90以上,較CN原料的研究法辛烷值(61.9)有了很大的提高。因此可以預測,進料中CN比例降低時,改質汽油辛烷值可進一步提高。在工業應用中,如果CN比例較大,可采用兩段提升管的耦合方式:一段提升管CGO與CN組合進料、二段提升管循環油與CN組合進料。

表10 CGO與CN組合進料汽油產品的研究法辛烷值
4 結 論
兩段提升管催化裂解多產丙烯技術對于CGO具有很好的適應性,實驗結果表明,在特定催化劑及工藝條件下,CGO具有較高的轉化率,可達到87.80%,而液化氣收率可以達到35%以上,丙烯收率為18.12%。利用組合進料技術,不僅可以對CN進行有效的改質,提高CN的辛烷值與穩定性,而且可以提高CGO的加工效率,為延遲焦化-催化裂化聯合工藝提供新的加工思路與加工路線。
[1] 袁晴棠.中國劣質原油加工技術進展與展望[J].當代石油化工,2007,15(12):1-6
[2] Mohan S Rana,Vicente Sámano,Jorge Ancheyta,et al.A review of recent advances on process technologies for upgrading of heavy oils and residual[J].Fuel,2007,86(9):1216-1231
[3] 劉曉欣.催化裂化-延遲焦化組合工藝[J].石油煉制與化工,1998,29(11):28-33
[4] 王雪松,袁志祥,尹魯江,等.延遲焦化工藝的技術進展[J].工業催化,2006,14(4):22-25
[5] 黃新龍,沙穎遜,曲賀欣,等.延遲焦化-溶劑精制-催化裂化組合工藝應用[J].煉油設計,2001,31(6):48-51
[6] 瞿國華,黃大智,梁文杰.延遲焦化在我國石油加工中的地位和前景[J].石油學報(石油加工),2005,21(3):47-53
[7] 陳文藝,欒錫林,關易達.我國焦化蠟油的組成和特性[J].石油化工,2000,29(8):607-612
[8] Cerqueira H S,Caeiro G,Costa L,et al.Deactivation of FCC catalysts[J].Journal of Molecular Catalysis A:Chemical,2008,292(1):1-13
[9] Li Chunyi,Yang Chaohe,Shan Honghong.Maximizing propylene yield by two-stage riser catalytic cracking of heavy oil[J].Ind Eng Chem Res,2007,46(14):4914-4920
[10]李曉紅.兩段提升管催化裂化多產丙烯(TMP)技術應用基礎研究[D].東營:中國石油大學(華東),2007
[11]李春義,徐占武,姜國驊,等.兩段提升管催化裂解多產丙烯技術的工業試驗[J].石化技術與應用,2008,26(5):436-441
[12]楊朝合,李春義,山紅紅,等.一種利用兩段催化裂解生產丙烯和高品質汽柴油的方法:中國,CN101074392[P].2007-11-21
[13]鐘樂,霍永清,王均華,等.常壓渣油多產液化氣和汽油(ARGG)工藝技術[J].石油煉制與化工,1996,26(6):15-19
[14]余本德,施至誠,許友好.CRP21裂解催化劑工業應用及15萬t/a催化裂解裝置開工運轉[J].石油煉制與化工,1995,26(5):7-13
[15]趙曉波.改性納米HZSM-5在低品質輕油改質中的應用研究[D].大連:大連理工大學,2006
[16]曹壽康,劉丹禾,余安平,等.劣質汽油催化改質——芳構化催化劑:中國,CN1023687[P].1994-01-05
[17]陳祖庇,張久順,鐘樂燊,等.MGD工藝技術的特點[J].石油煉制與化工,2002,33(3):21-25
[18]張國才.焦化汽油的催化裂化改質[J].石油煉制與化工,2001,32(4):5-9
[19]李成霞,張強,李春義,等.焦化汽油催化裂化改質的反應條件研究[J].煉油技術與工程,2005,35(6):11-14
[20]Attila Lengyel,Szabolcs Magyar,Jen?Hancsók.Upgrading of delayed coker light naphtha in a crude oil refinery[J].Petroleum &Coal,2009,51(2):80-90
[21]梁文杰,闕國和,劉晨光,等.石油化學[M].2版.東營:中國石油大學出版社,2009:330
[22]Torre I,Arandes J M,Azkoiti M J,et al.Cracking of coker naphtha with gas-oil:Effect of HZSM-5zeolite addition to the catalyst[J].Energy &Fuels,2007,21(1):11-18
[23]袁啟民.焦化蠟油催化轉化應用基礎研究[D].東營:中國石油大學(華東),2007
[24]劉銀東,李澤坤,王剛,等.競爭吸附對催化裂化反應過程的影響[J].化工學報,2008,59(11):2794-2799
[25]劉銀東,王剛,李澤坤,等.油劑混合狀態對焦化蠟油催化裂化反應的影響[J].煉油技術與工程,2009,39(5):8-11
[26]李春義,高傳承,司冠飛,等.利用催化裂解焦化蠟油及改質焦化石腦油的多產丙烯方法:中國,CN100910229606.1[P].2010-05-12
[27]Greensfelder B S,Voge H H,Good G M.Catalytic and thermal cracking of pure hydrocarbons[J].Industrial and Engineering Chemistry,1949,41(11):2573-2584
猜你喜歡
電子樂園·下旬刊(2022年5期)2022-05-13 20:42:21
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
石油石化綠色低碳(2019年6期)2019-01-14 01:16:16
石油石化綠色低碳(2019年6期)2019-01-14 01:16:14
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
當代化工研究(2016年6期)2016-03-20 16:21:37
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
化工進展(2015年6期)2015-11-13 00:26:37
