中藥中赭曲霉毒素A的測定
2011-01-25 09:35:02鄭榮簡龍海毛丹王少敏王柯季申
中成藥 2011年10期
鄭榮,簡龍海,毛丹,王少敏,王柯,季申
(上海市食品藥品檢驗所,上海201210)
赭曲霉毒素是赭色曲霉屬Ochraceors和幾種青霉屬真菌產(chǎn)生的一種毒素,包括A、B、C等7種結(jié)構(gòu)類似的化合物,其中赭曲霉毒素A(ochratoxin A)被公認為是目前致癌力最強的天然物質(zhì)之一。毒理學資料表明,這種致癌作用主要體現(xiàn)在肝臟與腎臟,并被認為與人類的巴爾干腎病有關(guān),國際癌癥研究機構(gòu)IARC將其定為2B類致癌物[1-2]。
目前對赭曲霉毒素的研究多集中在谷物、糧油等食品樣本中[3],各國針對糧食中的赭曲霉毒素也制定了嚴格的限度標準。而在與食品具有類似基質(zhì)的中藥中尚未建立相應的檢測方法與標準。2000年6月8日,美國《新英格蘭醫(yī)學雜志》報道了43位服用含有馬兜鈴屬中草藥及千金藤、玉木蘭減肥藥而出現(xiàn)嚴重腎功能衰竭,其中4例有赭曲霉毒素(存在于千金藤)中毒的證據(jù)。為此,建立中藥中赭曲霉毒素A的檢測方法,不僅可以確保人民用藥安全,也可以為監(jiān)督執(zhí)法提供強有力的技術(shù)支撐。
目前文獻報道的赭曲霉毒素A的分析測定方法有薄層色譜法(TLC)、酶聯(lián)吸附免疫法(ELISA)和高效液相色譜法(HPLC)[4-12]。,由于中藥基質(zhì)復雜,TLC法和ELISA法受方法本身的限制,假陽性率較高。經(jīng)實驗摸索研究,建立了中藥中免疫親和凈化-高效液相色譜的測定方法,陽性樣品采用液相色譜-串聯(lián)質(zhì)譜進行確證。該方法快速靈敏,回收率高、準確度好、可作為中藥中赭曲霉毒素A的測定方法。
1 儀器與試藥
高速均質(zhì)器(德國IKA公司);赭曲霉毒素A免疫親和柱(美國VICAM公司);美國Agilent 1100高效液相色譜儀,熒光檢測器,赭曲霉毒素A對照品(Sigma公司提供,批號:126K4026)。
香豆豉、防己、薏苡仁、補骨脂、麥芽、稻芽、白扁豆、綠豆、赤小豆藥材均由華宇藥材有限公司提供,經(jīng)上海市食品藥品檢驗所鑒定。
2 方法與結(jié)果
2.1 色譜條件用十八烷基硅烷鍵合硅膠為填充劑;乙腈-2%冰乙酸水溶液(49∶51)為流動相,體積流量1.0 mL/min;以熒光檢測器檢測,激發(fā)波長333 nm,發(fā)射波長477 nm。進樣量:10 μL。理論板數(shù)以赭曲霉毒素A計應不低于4 000。對照品溶液及陽性樣品的色譜圖見圖1~2。
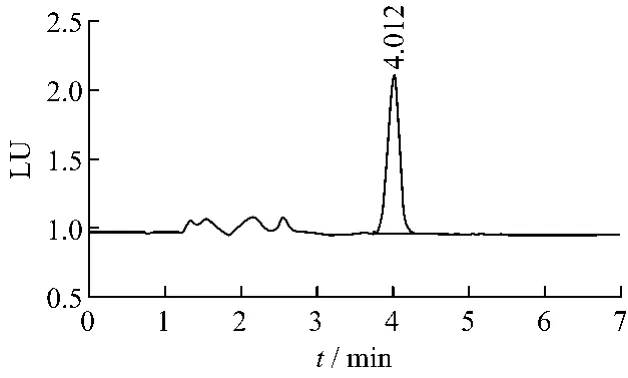
圖1 赭曲霉毒素A對照品的液相色譜圖Fig.1 HPLC chromatogram of ochratoxin A standard
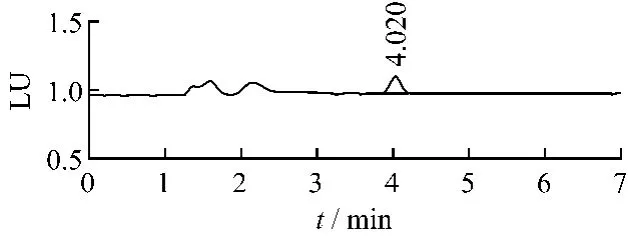
圖2 陽性樣品的液相色譜圖Fig.2 HPLC chromatogram of ochratoxin A contaminated sample
2.2 供試品溶液的制備取樣品粉末20g(過二號篩),精密稱定,加入氯化鈉5 g,80%甲醇100 mL,高速攪拌2 min,離心5 min(離心速度2 500 r/min),精密吸取上清液10 mL,用水稀釋至50 mL,搖勻,濾過,取續(xù)濾液10 mL,通過免疫親合柱(體積流量3 mL/min.),隨后用水20 mL洗脫(體積流量6 mL/min.,必要時可以先用10 mL淋洗緩沖液(稱取25 g氯化鈉、5 g碳酸氫鈉溶于水中,加入0.1 mL吐溫-20,用水稀釋至1 L,即可)洗脫,再用10 mL水洗脫),洗脫液棄去,精密量取1 mL甲醇洗脫(體積流量1 mL/min.),收集甲醇洗脫液,即得。
2.3 線性關(guān)系精密稱取赭曲霉毒素A對照品適量,用甲醇配制成質(zhì)量濃度為0.1 mg/mL的赭曲霉毒素A對照品貯備液(4℃冰箱保存)。分別精密吸取對照品貯備液適量,用甲醇稀釋制得質(zhì)量濃度分別為1、2、5、10、15、20 ng/mL的對照品溶液。精密吸取上述對照品溶液各10 μL,注入液相色譜儀,以濃度為橫坐標,峰面積為縱坐標進行回歸計算。結(jié)果赭曲霉毒素A的回歸方程為Y=0.229X+0.049 4,r=0.999 94。表明赭曲霉毒素A在1~20 ng/mL濃度范圍內(nèi)線性關(guān)系良好。
2.4 進樣精密度試驗取質(zhì)量濃度為2 ng/mL的對照品溶液,連續(xù)進樣6次,結(jié)果表明,進樣精密度為0.5%。
2.5 重復性試驗分別取薏苡仁及麥芽的陽性樣品粉末(過二號篩)20 g,一式6份,精密稱定,按供試品溶液的制備項下依法操作,進樣分析,計算RSD值分別為6.6%和9.7%,表明重復性試驗結(jié)果良好。
供試品溶液采用LC-MS/MS進行陽性確證,結(jié)果(見表1)供試品溶液中定性離子的豐度比與對照品溶液中定性離子的豐度比相接近(偏差均小于15%)。
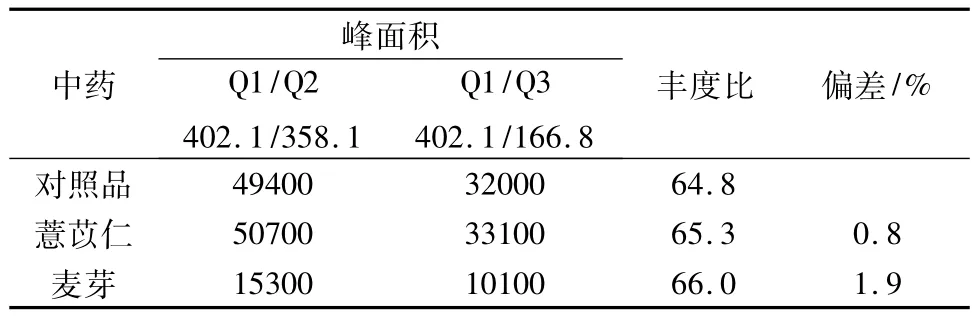
表1 質(zhì)譜確證結(jié)果Tab.1 LC-MS/MS confirmation results
2.6 回收率試驗取樣品粉末(過二號篩)20 g,精密稱定,一式12份,分別精密加入赭曲霉毒素A對照品50 ng、80 ng及300 ng,做為低、中、高三種濃度添加水平的供試品溶液,測定,計算得平均回收率(見表2),結(jié)果表明本方法回收率試驗結(jié)果良好。
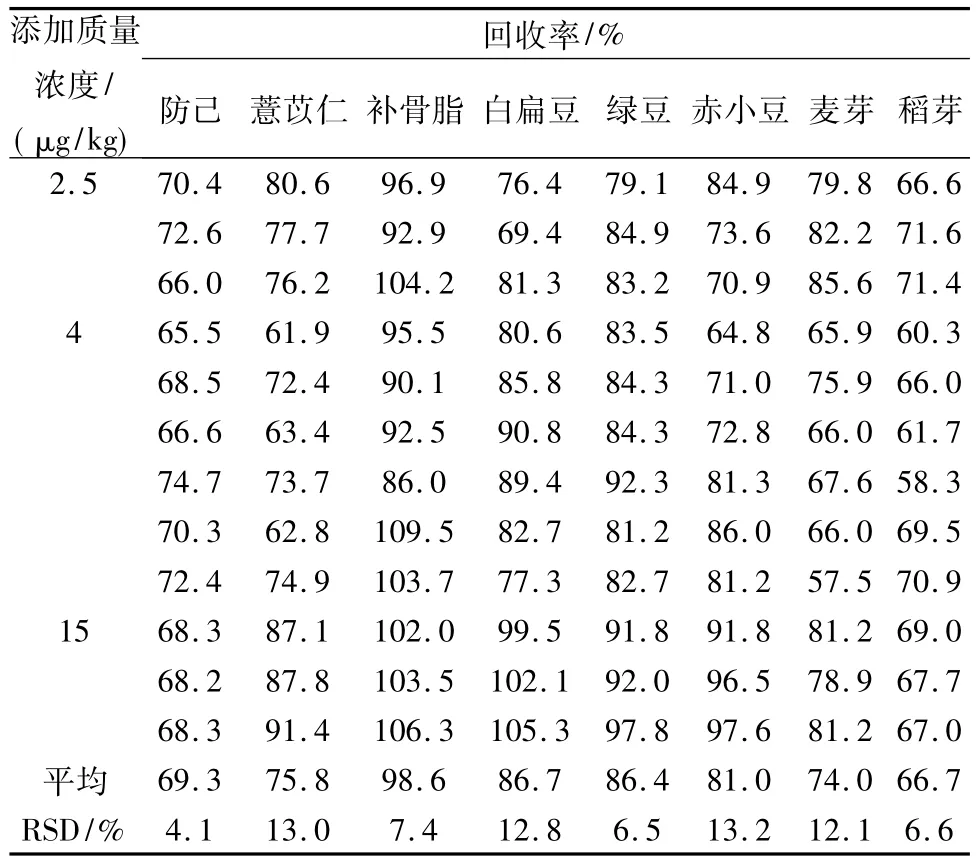
表2 回收率試驗結(jié)果Tab.2 Results of recovery
2.7 穩(wěn)定性試驗取回收率試驗項下中濃度添加水平的樣品,每隔4 h進樣分析,記錄測得量(單位:pg),見表3,結(jié)果表明,在0~24 h之內(nèi),供試品溶液保持穩(wěn)定。
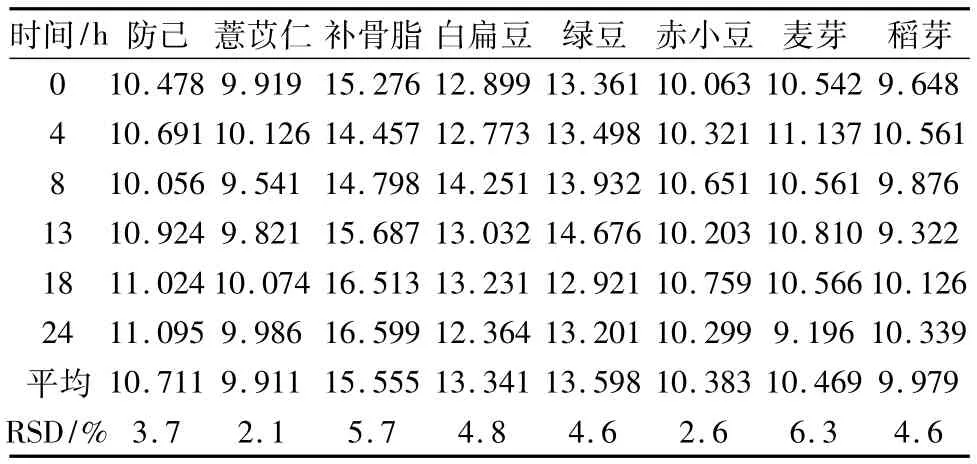
表3 穩(wěn)定性試驗結(jié)果Tab.3 Results of stablity/pg
2.8 檢測限測定低濃度回收率供試品溶液的信噪比,以信噪比3∶1計算得本方法檢測限為1 μg/kg,以信噪比10∶1計算得本方法定量限為3 μg/kg。
2.9 樣品測定
2.9.1 國際實驗室能力測試結(jié)果按照本實驗所述方法參加由英國實驗室CSL主辦的能力驗證測試項目,一式3份的結(jié)果分別為6.32 μg/kg、6.14 μg/kg、6.10 μg/kg,與中位值為6.16 μg/kg接近。2.9.2樣品測定結(jié)果對上述8種共計21批樣品進行檢驗,結(jié)果1批薏苡仁檢出赭曲霉毒素A(質(zhì)量分數(shù)為5 μg/kg),1批麥芽檢出赭曲霉毒素A(質(zhì)量分數(shù)為1 μg/kg)。
2.10 確證實驗供試品溶液制備方法中使用的免疫親和柱對赭曲霉毒素具有專屬性吸附,因此一般情況下供試品溶液的高效液相色譜圖比較干凈,出現(xiàn)假陽性的情況比較少。筆者在近幾年的實驗中發(fā)現(xiàn),隨著監(jiān)測品種范圍和檢驗樣品數(shù)量的擴大,某些基質(zhì)復雜的樣品,有時會出現(xiàn)假陽性的情況。因此進一步的確證實驗就顯得非常必要。通常,建議通過改變色譜條件比如更換儀器和色譜柱,調(diào)節(jié)流動相比例等手段進行考察。有條件的實驗室,則應通過LC-MS進行確證。
本實驗采用LC-MS-MS對上述陽性樣品進行了確證實驗。色譜柱為ACQUITY UPLC BEH C18(1.7 μm,2.1 mm×100 mm);以甲醇為流動相A相,以0.01%甲酸為流動相B相,體積流量0.3 mL/min;按表4進行梯度洗脫。
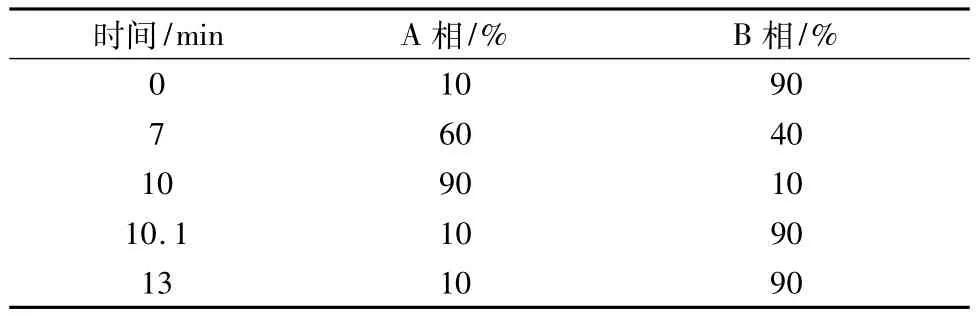
表4 流動相梯度Tab.4 Mobile phase gradient program
離子源為電噴霧離子化源ESI(-)源;毛細管去簇電壓、碰撞池能量等質(zhì)譜參數(shù)見表5。
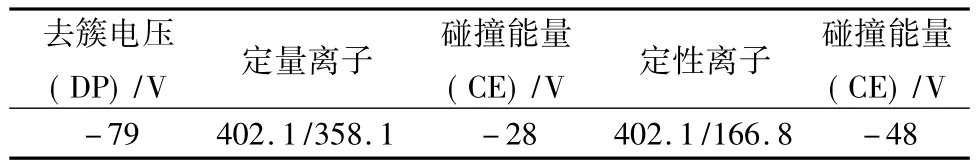
表5 質(zhì)譜參數(shù)表Tab.5 LC-MS/MS parameters
3 討論
3.1 色譜條件與系統(tǒng)適用性試驗
3.1.1 波長的選擇分別考察了①激發(fā)波長360 nm,發(fā)射波長477 nm,②激發(fā)波長333 nm,發(fā)射波長477 nm,③激發(fā)波長333 nm,發(fā)射波長460 nm,④激發(fā)波長333 nm,發(fā)射波長420 nm,⑤激發(fā)波長333 nm,發(fā)射波長440 nm。結(jié)果以激發(fā)波長333 nm,發(fā)射波長477 nm所得色譜峰峰面積最大,峰高最高。
3.1.2 流動相系統(tǒng)的選擇分別考察了①乙腈-水(40∶60)、②乙腈-0.012 mol/L磷酸鈉溶液pH7.5(60∶40),1 g/L十六烷基三甲基溴酸銨、③乙腈-2%冰醋酸(49∶51)3種流動相,對稱因子分別為0.75、0.81、0.84。故選用對稱因子較好且配制比較方便的乙腈-2%冰醋酸(49∶51)為流動相。
3.1.3 色譜柱的選擇分別考察了①INTERSILODS 3(5 μm,4.6 mm×150 mm)、②Agilent CB-C18(5 μm,4.6 mm×250 mm)、③Waters C18(5 μm,2.1 mm×150 mm)3種色譜柱,對稱因子分別為0.81、0.83、0.76。色譜柱①及色譜柱②的對稱因子相對為好。
3.2 供試品溶液制備方法的考察
3.2.1 提取過程選用目前比較先進的前處理手段進行了不同提取過程的比較。取樣品粉末20 g(過二號篩),分別考察了表6所述的提取溶劑及提取方式。結(jié)果表明,采用80%甲醇勻漿1 min即可提取完全,為保證提取效率,確定勻漿時間為2 min。
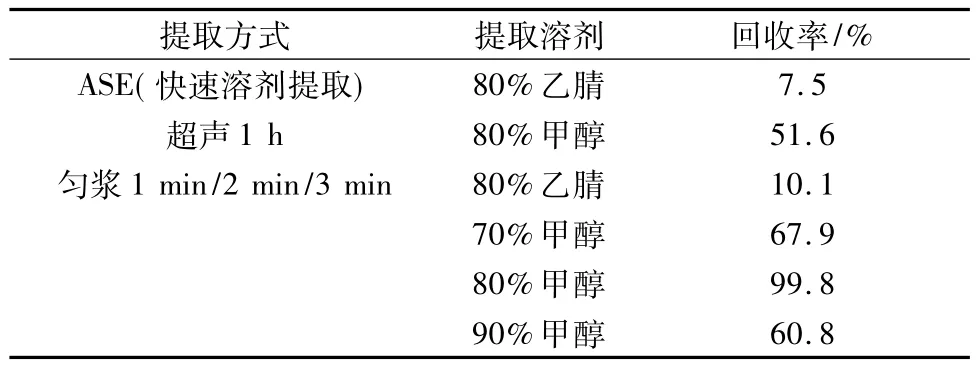
表6 不同提取溶劑及提取方式的比較Tab.6 Comparison of different extraction solvents and preparation methods
3.2.2 凈化過程分別取供試品溶液制備項下的續(xù)濾液10 mL、20 mL進行凈化,結(jié)果表明上柱體積不影響測定結(jié)果,因此在不超過柱容量的前提下,可以通過增加上柱體積來提高供試品溶液中赭曲霉毒素A的濃度。
對淋洗液種類(淋洗緩沖液和水)及用量的考察結(jié)果表明,用20 mL水即可以去除大部分的雜質(zhì)干擾,因此,一般情況下不需要使用淋洗緩沖液,樣品中的干擾雜質(zhì)特別多的情況下可以先用10 mL淋洗緩沖液洗脫,再用10 mL水洗脫。
甲醇用量考察結(jié)果表明1 mL甲醇即可洗脫完全,增大甲醇用量會降低供試品溶液中赭曲霉毒素A的濃度,影響檢測靈敏度,為此確定甲醇的洗脫用量為1 mL。
3.2.3 濃縮過程將供試品溶液制備項下的甲醇洗脫液一份氮吹濃縮后進樣分析,一份直接進樣分析。結(jié)果表明,氮吹不會造成赭曲霉毒素A的損失。因此,若供試品中赭曲霉毒素A的量過低,可以通過氮吹進行濃縮,提高樣品中赭曲霉毒素A的濃度。
在不超出免疫親和柱最大載樣量的情況下,可以通過改變樣品稱樣量、加水量以及上柱體積,或氮吹等步驟來調(diào)整供試品溶液中赭曲霉毒素A的濃度,以達到良好的液相分離效果。
3.3 中藥中赭曲霉毒素的限量目前國內(nèi)外暫無中藥中赭曲霉毒素A的限量標準。國際食品法典委員會提議赭曲霉毒素A在小麥、大麥、黑麥及其它們的制品中不得超過5 μg/kg。瑞士規(guī)定豬混合料中限度為200 μg/kg,家禽混合料中限度為1000 μg/kg。歐盟規(guī)定速溶咖啡中限度為6 μg/kg,烤制咖啡中限度為3 μg/kg,干果中限度為2 μg/kg,干無花果中限度為8 μg/kg。德國規(guī)定咖啡及其制品中限度為5 μg/kg。目前我國規(guī)定谷類食品中赭曲霉毒素A的限量標準為5 μg/kg[13-14]。據(jù)此,認為可暫參照食品標準規(guī)定中藥中赭曲霉毒素A的限度為不得過5 μg/kg。
[1]張藝兵,鮑蕾,褚慶華.農(nóng)產(chǎn)品中真菌毒素的檢測分析[M].北京:化學工業(yè)出版社,2006.
[2]Schlatter C,Studer-Rohr J,Rasonyi T.Carcinogenicity and kinetic aspects of ochratoxin A[J].Food Addit Contam,1996,13(Suppl):43-44.
[3]蔡梅,曹玉潔,嚴小平.玉米、小麥中赭曲霉毒素A污染情況調(diào)查[J].江蘇預防醫(yī)學,1994,5(1):47-48.
[4]Entwisle A C,Williams A C,Mann P J,et al.Liquid chromatographic method with immunoaffinity column cleanup for determination of ochratoxin A in barley:collaborative study[J].J Aoac Int,2000,83(6):1377-1383.
[5]GB/T23502-2009.食品中赭曲霉毒素A的測定-免疫親和層析凈化高效液相色譜法[S].
[6]GB/T 5009.96-2003.谷物和大豆中赭曲霉毒素A的測定[S].
[7]時瑾,黃飚,孫蔚榕,等.ELISA法檢測赭曲霉毒素A的改進研究[J].食品科學,2007,28(8):425-428.
[8]SN/T1746-2006.進出口大豆、油菜籽和食用植物油中赭曲霉毒素A的檢驗方法[S].
[9]SN/T1940-2007.進出口食品中赭曲霉毒素A的測定方法[S].
[10]Sibanda L,De Saeger S,Van Peteghem C.Optimization of solidphase clean-up prior to liquid chromatographic analysis of ochratoxin A in roasted coffee[J].J Chromatogr A.2002,959(1-2):327.
[11]Entwisle A C,Williams A C,Mann P J,et al.Combined phenyl silane and immunoaffinity column cleanup with liquid chromatography for determination of ochratoxin A in roasted coffee:collaborative study[J].J Aoac Int,2001,84(2):444.
[12]樊祥,褚慶華,周瑤,等.反相高效液相色譜法測定張家口赤城大麥中赭曲霉毒素A的含量[J].分析試驗室,2007(S1):359-361.
[13]GB2715-2005.食品中真菌毒素限量[S].
[14]GB2761-2005.食品中真菌毒素限量[S].
