關于藥品再注冊注射劑研究資料審評標準的探討
2011-03-06 08:46:54段春改曹文利趙晨陽張偉琳
河北醫藥 2011年13期
段春改 曹文利 趙晨陽 張偉琳
為加強藥品注冊管理,保障人體用藥安全,按照國務院整頓和規范藥品市場秩序專項行動的統一部署,自2007年起,國家局擬對核發的藥品批準文號有效期滿5年,需要繼續生產的藥品啟動再注冊工作。目前再注冊的集中審評已經結束,成為日常性工作,但是在審評過程中遇到許多值得我們共同探討的問題,尤其是安全性風險比較大的注射劑研究資料的審評工作,更值得我們共同商榷。根據安排,各省級藥品監管部門將依照《藥品管理法》等有關法律法規,結合藥品批準文號清查、藥品生產工藝和處方核查結果,開展藥品再注冊審查工作。河北省審評認證中心會同省局注冊處、安監處、市場處、省藥檢所以及省內大型制藥企業包括石藥集團、華藥集團和石家莊四藥股份有限公司等先后召開專題研討會或座談會,并到企業中調研,最終綜合考慮各方面要求,按照《化學藥品注射劑基本技術要求(試行)》和《多組分生化藥注射劑基本技術要求(試行)》(以下簡稱注射劑基本技術要求)進行注射劑研究資料的審評。
1 充分借鑒和依據前期工作
根據國家局要求,綜合幾次會議精神,以及調研情況,藥品再注冊審查工作充分結合藥品批準文號清查、藥品生產工藝和處方核查結果開展。注射劑的藥品生產工藝和處方核查(以下簡稱工藝核查)是注射劑審查的一項必要依據,要求資料中必須附安監處的核查結果。核查結果為“繼續生產”,予以再注冊;核查結果為“繼續生產,限期申報”,企業應提供申報補充申請的相關資料,予以再注冊。未進行工藝核查,根據國家局在隨后的【2010】168號發函,對未進行工藝核查或核查結果為“責令停產”的,待核查或復查符合要求后,予以再注冊。在審評過程發現,企業容易缺失限期申報要求完成工作的資料,如原料供應商的變更等。工藝核查是注射劑審查的一項必要依據。
2 區分再注冊與新藥注冊的不同
依據注射劑基本技術要求,充分考慮再注冊不同于新藥注冊,作為老品種各方面技術已經比較成熟,所附研究資料與新藥的要求有很大區別,一般要求做綜述性陳述,或說明目前控制保障狀態。審查的要點為:劑型、規格選擇的必要性、合理性;注射劑原、輔料質量控制及來源;注射劑處方及制備工藝合理性、可行性研究,特別是滅菌工藝的驗證研究等;注射劑質量研究;注射劑穩定性研究。
2.1 劑型、規格的合理性、必要性 此項不再要求提供完整的藥學、藥理以及臨床研究資料,只要求綜述性陳述本品種的劑型、規格的合理性和必要性。列出該品種劑型、規格,根據說明書中規定的用法用量,從方便臨床用藥、滿足臨床用藥需要的角度加以闡述說明。但是一些企業卻容易漏失,沒有此項內容,直接從工藝驗證資料開始。
2.2 原、輔料藥質量控制及來源 本項需要按照相應項下要求說明目前的控制和保障狀態,提供原、輔料藥合法來源及質量控制的詳細資料,如果有原料來源變更請附相應補充申請。(1)原料藥質量控制:采用新研制的原料藥與制劑同時申報的,應按照注射用原料藥的要求提供規范完整的申報研究資料。有注射用原料藥上市的,應使用有合法來源的注射用原料藥。提供原料藥合法來源及質量控制的詳細資料,包括生產企業、批準文號、執行的質量標準等。若為進口原料藥,還應提供進口注冊證。若尚無注射用原料藥上市,需對原料藥進行精制并制定內控標準,使其達到注射用的質量要求。(2)輔料質量控制:使用已批準上市的注射用輔料,應提供輔料來源及質量控制的詳細資料,包括生產企業、執行的質量標準等,有批準文號的還應提供批準文號,進口輔料還應提供進口注冊證。使用尚未批準供注射途經使用的輔料,對于注射劑中有使用依據,但尚無符合注射用標準產品生產或進口的輔料,可對非注射途經輔料進行精制使其符合注射用要求,并制定內控標準。
需要特別說明的是,如果尚無注射用原料藥上市,需對原料藥進行精制并制定內控標準,使其達到注射用的質量要求。在注冊申請時,除提供相關的證明性文件外,應提供精制的工藝驗證資料和質量控制資料。如本省的注射用維生素C就屬于此類情況。
2.3 注射劑處方及制備工藝合理性、可行性研究,特別是滅菌工藝驗證等。
處方組成:列出處方中主藥及輔料成份,并寫明各種輔料在處方中的作用,并分析輔料用量及與主藥的相互作用。
制備工藝驗證項要求提供工藝流程圖(標明潔凈區級別)、工藝描述、工藝驗證資料、滅菌工藝驗證資料。提供完整的制備工藝及工藝驗證的結果,對工藝過程本身是否穩定,是否易于控制進行總結和評價。需要注意需要混粉的品種,在處方組成、工藝描述以及工藝驗證中都要體現混粉并進行相應驗證。
滅菌工藝驗證工作可結合生產線驗證一并進行。不同劑型滅菌工藝驗證資料也不同。
大容量注射劑需要準備終端滅菌工藝驗證資料。終端滅菌產品,滅菌條件一般達到F0≥8,均應保證產品滅菌后的SAL不大于10-6。滅菌工藝驗證主要包括以下試驗:滅菌前微生物污染水平測定,包括滅菌前產品中的污染菌及其耐熱性的測定;熱穿透試驗;微生物挑戰試驗:所用生物指示劑的耐熱性及數量應對滅菌工藝構成必要的挑戰,生物指示劑的耐熱性應大于產品中常見污染菌的耐熱性。采用過度殺滅法(F0≥12)滅菌工藝的,可不進行微生物挑戰試驗。
凍干粉針劑常規工藝驗證包括培養基模擬灌裝驗證和除菌過濾系統適應性驗證資料。培養基模擬灌裝驗證試驗:至少在線灌裝三批,每批的批量詳見表1,判斷該試驗是否合格的標準見表1。應進行陽性對照試驗及陰性對照試驗。除菌過濾系統適應性驗證試驗:包括過濾系統相容性測試、過濾前后濾膜完整性測試,必要時尚需進行濾膜的微生物截留量測試。無菌分裝粉針劑工藝驗證工作主要為培養基灌裝驗證試驗。灌裝的批數、批量與合格標準見表1。
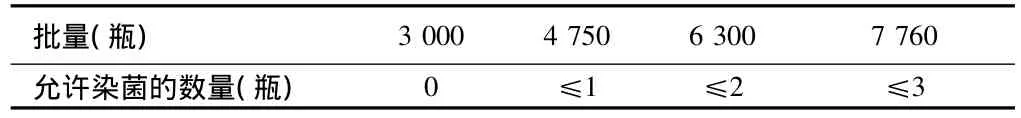
表1 培養基灌裝試驗的批量與判斷合格的標準
小容量注射劑滅菌工藝驗證資料因工藝的區別與大容量注射劑或凍干粉針的要求一致。需要注意的是,國家局在《關于做好藥品再注冊審查審批工作的補充通知》食藥監注【2010】394號中,進一步加強了無菌保障水平F0值﹤8的品種無菌保障,要求應至少提交無菌工藝驗證研究資料。因此這些品種如有充分的依據證明不能采用終端滅菌工藝,且為臨床必需注射給藥的品種,可考慮采用無菌生產工藝,相關技術要求同凍干粉針劑。對于過濾除菌工藝同時采用了流通蒸汽輔助滅菌的品種,建議修改為終端滅菌工藝,技術要求同大容量注射劑;對確實無法采用終端滅菌工藝的品種,應修改為無菌生產工藝,技術要求同凍干粉針劑。對于采用無菌生產工藝生產的小容量注射劑,生產線的驗證應結合無菌生產工藝進行。
生化藥參照以上相應劑型進行。注射劑基本技術要求特別提到,由于生化藥的特殊性,需關注滅菌處理對產品質量/療效以及產品安全性的影響。生化藥的原材料來源于不同的動物組織或者體液,為保持特定/有效成分的生物活性,通常需要采用比較溫和的生產工藝和提取制備條件,因而污染的潛在病毒可能未得到有效滅活/去除,給用藥人群帶來感染的風險性。因此,必須確立在生產工藝中包含能夠有效滅活/去除病毒的特定工藝步驟,并驗證其滅活/去除病毒的效能,提供病毒滅活/去除有效性驗證資料。以保證制品的病毒安全性。
2.4 化學藥品注射劑質量研究 請附現執行質量標準;檢驗方法驗證至少應提供無菌檢驗方法的驗證。
2.5 化學藥品注射劑穩定性研究 注射劑穩定性研究的樣品批次和規模、包裝和放置條件和考察時間點參見《化學藥物穩定性研究技術指導原則》,考察項目通常應包括性狀、pH值/酸堿度、澄清度與顏色(或溶液的澄清度與顏色)、有關物質、干燥失重/水分(注射用粉末)、細菌內毒素/熱原、無菌、不溶性微粒、可見異物、含量(效價)測定。注射劑穩定性研究內容包括影響因素試驗、加速試驗和長期試驗,具體內容參見《化學藥物穩定性研究技術指導原則》。報告中應包括穩定性研究結果評價。
3 專家審評把關注射劑審評
為了進一步做好注射劑的再注冊工作,經過分析研究,化學藥品和多組分化藥注射劑需要通過組織專家技術審評通過后,方予以注冊。審評專家從專家庫中隨機抽取決定。專家庫由本省科研單位、藥品檢驗單位、疾病控制中心以及省內大型生產單位的學術帶頭人組成。以此保證了注射劑審評質量。
總之,藥品再注冊是一項重要工作任務,當前我國藥品標準水平還不能完全適應監管工作的需要,藥監部門將把黨中央提出的“用最嚴格的藥品標準監督藥品安全”的要求落實到具體工作中,圍繞提高藥品質量、保障用藥安全的總體要求,進一步規范藥品審評行為,嚴把藥品審批關。
猜你喜歡
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
山東冶金(2019年6期)2020-01-06 07:45:54
世界農藥(2019年2期)2019-07-13 05:55:12
小學生優秀作文(低年級)(2018年6期)2018-05-19 01:54:28
中國衛生(2016年5期)2016-11-12 13:25:28
銅業工程(2015年4期)2015-12-29 02:48:39
中國衛生(2015年9期)2015-11-10 03:11:14
中國衛生(2015年5期)2015-11-08 12:09:48
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
中國衛生(2014年7期)2014-11-10 02:33:02
