參芪二仙片質量標準研究
2011-07-27 07:09:00趙玉華崔曉娟王衛鋒石敏娟
中國醫藥導報 2011年31期
趙玉華 ,崔曉娟 ,樊 萍 ,王衛鋒 ,李 芳 ,石敏娟
1.陜西中醫學院藥學院,陜西 咸陽 712046;2.陜西省中醫藥研究院,陜西 西安 710003
參芪二仙片為中華人民共和國衛生部 《中藥成方制劑》第20冊中所收載,是由紅參、黃芪、當歸等八味中藥組成的中藥復方制劑,具有補腎填精、調補沖任、益氣養血的功效,用于腎虛腰膝酸軟、陽痿早泄、遺精、婦女更年期經血不調等癥。原質量標準中,僅有一項顯微鑒別,為了更好地控制該制劑的質量,參照有關文獻[1-5],筆者對方中的主要藥味,進行了專屬性較強的薄層鑒別研究,并對方中的主要藥物淫羊藿中的淫羊藿苷進行了含量測定的方法學研究,結果此方法具有分離效果好、靈敏、準確等優點。現將結果報道如下:
1 儀器與試藥
高效液相色譜儀 (美國SSI)、超聲波清洗機 (KH-300DE)、BP211D型分析天平 (德國賽多利斯)、TGL-16G離心機(上海安亭科學儀器廠);人參皂苷Rg1對照品(中國藥品生物制品檢定所,批號:110703-200726,供含量測定用)、人參皂苷Rb1對照品 (中國藥品生物制品檢定所,批號:110704-200420,供含量測定用)、當歸對照藥材(中國藥品生物制品檢定所,批號:120927-200613,供鑒別用)、鹽酸小檗堿對照品(中國藥品生物制品檢定所,批號:100713-200609,供含量測定用)、淫羊藿苷對照品 (中國藥品生物制品檢定所,批號:110737-200415,供含量測定用)。參芪二仙片自制(批號:080501、080502、080503); 乙腈為色譜純, 水為超純水,其他試劑均為分析純。
2 方法與結果
2.1 薄層色譜鑒別
2.1.1 TLC法鑒別紅參 取本品25片,除去包衣,研細,置索氏提取器中,加三氯甲烷回流提取3 h,三氯甲烷液備用;藥渣繼續用甲醇回流提取至近無色,提取液回收甲醇并濃縮至干,殘渣加水20 ml使溶解,用水飽和正丁醇振搖提取3次,每次20 ml,合并正丁醇液,用氨試液洗滌2次,每次20 ml,棄去,再用水洗2次,每次20 ml,棄去,正丁醇液水浴蒸干,殘渣加水5 ml使溶解,然后上處理好的D101大孔吸附樹脂柱(直徑 1.5 cm,高12 cm),先用水150 ml洗脫,棄去,再用50%乙醇50 ml洗脫,收集洗脫液,水浴蒸干,殘渣加甲醇2 ml使溶解,作為供試品溶液。另取人參皂苷Rg1和Rb1對照品,加甲醇制成每毫升含1 mg的混合溶液,作為對照品溶液。參照薄層色譜法(《中國藥典》2010年版一部附錄Ⅵ B)試驗,吸取上述兩種溶液各5 μl,分別點于同一以羧甲基纖維素鈉為粘合劑的硅膠G薄層板上,以三氯甲烷-甲醇-水(13∶7∶2)10℃以下放置過夜后的下層溶液為展開劑,展開,取出,晾干,噴以10%硫酸乙醇溶液,105℃加熱約10 min使斑點顯色清晰。供試品色譜中,在與對照品色譜相應的位置上,顯相同顏色的斑點,且陰性對照無干擾。
2.1.2 TLC法鑒別當歸 取“2.1.1”項下的三氯甲烷提取液,水浴蒸至約5 ml作為供試品溶液。另取當歸對照藥材1 g,加乙醚20 ml回流提取30 min,濾過,濾液揮干,殘渣加乙酸乙酯1 ml使溶解作為對照藥材溶液。參照薄層色譜法(《中國藥典》2010年版一部附錄ⅥB)試驗,吸取上述兩種溶液各5 μl,分別點于同一以羧甲基纖維素鈉為粘合劑的硅膠G薄層板上,以正己烷-乙酸乙酯(9∶1)為展開劑,展開,取出,晾干,置紫外光燈(365 nm)下檢視,供試品色譜中,在與對照品色譜相應的位置上,顯相同顏色的熒光斑點,且陰性對照無干擾。
2.1.3 TLC法鑒別黃柏 取本品3片,除去包衣,研細,加甲醇20 ml回流提取15 min,濾過,濾液濃縮至5 ml,作為供試品溶液。另取鹽酸小檗堿對照品適量,加甲醇制成每毫升含0.5 mg的溶液作為對照品溶液;再取黃柏對照藥材0.5 g,同法制成對照藥材溶液。照薄層色譜法(《中國藥典》2010年版一部附錄ⅥB)試驗,吸取對照品溶液及對照藥材溶液各2 μl,供試品溶液5 μl,分別點于同一以羧甲基纖維素鈉為粘合劑的硅膠G薄層板上,以甲苯-乙酸乙酯-異丙醇-甲醇-濃氨(6.0∶3.0∶1.5∶1.5∶0.5)為展開劑,用氨蒸氣飽和后展開,取出,晾干,置紫外光燈(365 nm)下檢視,供試品色譜中,在與對照品色譜及對照藥材色譜相應的位置上,顯相同顏色的熒光斑點,且陰性對照無干擾。
2.2 淫羊藿苷含量測定
2.2.1 色譜條件與系統適用性試驗 色譜柱:Phenomenex ODS柱(150 mm ×4.6 mm,5 μm);流動相:乙腈-水(28∶72);流速:1.0 ml/min;檢測波長:270 nm;進樣量:5 μl。理論板數按淫羊藿苷峰計算應不低于2000。
2.2.2 對照品溶液的制備 精密稱取淫羊藿苷對照品適量,加甲醇制成每毫升含0.010 mg的淫羊藿苷對照品溶液,作為對照品溶液。
2.2.3 供試品溶液的制備 取樣品20片,精密稱定,除去包衣,研細,取約0.5 g,精密稱定,置50 ml量瓶中,加稀乙醇約45 ml,超聲處理30 min,放冷,加稀乙醇至刻度,搖勻,濾過,即得供試品溶液。
2.2.4 陰性對照品溶液的制備 取缺淫羊藿的陰性對照樣品,同供試品溶液的制備方法,制備成陰性對照溶液。
2.2.5 干擾試驗 照“2.2.1”項下色譜條件,吸取對照品溶液、供試品溶液、陰性對照溶液各5 μl,分別依次進樣。供試品色譜中,在與對照品色譜相應的位置上,有色譜峰出現,而陰性對照液在淫羊藿苷色譜峰位置處無相應峰出現,表明陰性無干擾。見圖1。
2.2.6 線性關系考察 取對照品溶液 (0.011 mg/ml)2、4、6、8、10、12 μl進樣,記錄色譜圖,測定峰面積,以峰面積(Y)對進樣量(X)進行回歸,得標準曲線方程 Y=2677350X-436,相關系數r=0.9999(n=6)。結果表明,淫羊藿苷進樣量在0.022~0.110 μg范圍內與峰面積呈良好的線性關系。
2.2.7 精密度試驗 取對照品溶液,按擬定的色譜條件測定,重復進樣6次,結果峰面積的RSD為1.89%(n=6)。表明儀器精密度良好。
2.2.8 穩定性試驗 取同一批號的供試液,分別每隔2 h進樣一次,于8 h內依法進樣,結果峰面積的RSD為1.96%(n=5)。表明供試品溶液在8 h內穩定。
2.2.9 重復性試驗 按擬定的含量測定方法,對同一樣品分別制備6份供試品溶液,測淫羊藿苷的含量,結果含量的RSD為1.51%(n=6)。結果表明方法重復性良好。
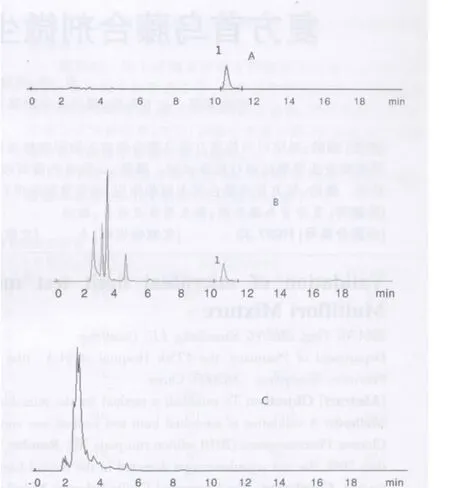
圖1 淫羊藿苷對照品及樣品色譜圖
2.2.10 加樣回收試驗 取已知含量的樣品 (淫羊藿苷含量:0.453 mg/片)6份,每份 0.25 g,精密稱定,分別加入濃度為0.11 mg/ml的淫羊藿苷對照品溶液 3 ml,按“2.2.3”項下操作,依法制備,進樣,測定峰面積,按公式1計算回收率。結果見表 1。
公式 1:回收率(%)=[測得量(mg)-制劑中含量(mg)]/加入對照品量(mg)×100%
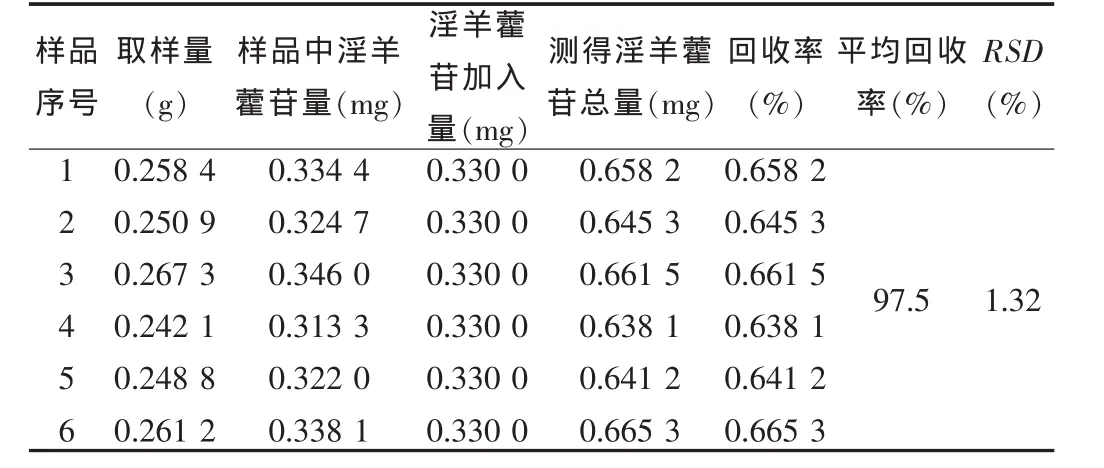
表1 回收率試驗結果
2.2.11 含量測定 取參芪二仙片三批樣品,按“2.2.3”項下操作制備供試品溶液,按“2.2.1”項下色譜條件依法測定。結果見表2。
3 討論
本制劑中,對紅參中的人參皂苷Re也進行了鑒別研究,結果斑點顯色模糊,因此未納入標準正文;含量測定中對樣品的提取方法進行了對比研究,結果顯示回流提取與超聲提取無本質區別,因此選擇較為簡單的超聲提取進行處理;提取溶劑選擇了甲醇、50%甲醇、乙醇和稀乙醇進行比較,結果稀乙醇提取效果較好。從樣品的含量測定數據可知,平均含量為0.447 mg/片,從大生產及藥材來源的角度考慮,暫定本品含淫羊藿苷不得低于0.350 mg/片。最終的含量測定限度,應繼續積累數據,再行確定。
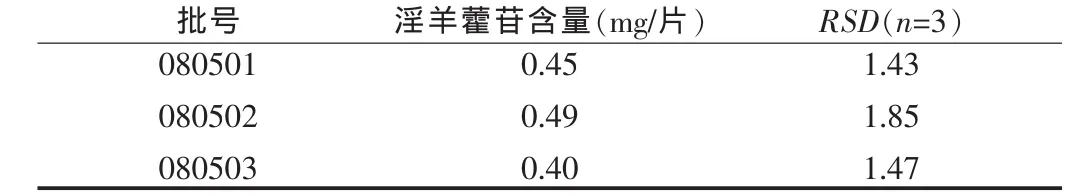
表2 參芪二仙片中淫羊藿苷的含量測定結果
[1]孫棟梁,黃慧成,熊國營.腎康寶膠囊質量標準研究[J].江西中醫藥,2007,38(5):66-67.
[2]湯磊,郭宗華.骨力膠囊質量標準研究[J].中成藥,2007,29(1):150-151.
[3]羅毅,楊俊,馬俊玲.淫黃顆粒質量控制方法研究[J].中國藥房,2007,18(3):200-201.
[4]安軍永,劉敏彥,王玉峰,等.高效液相色譜法測定仙靈骨葆顆粒中淫羊藿苷的含量[J].河北中醫藥學報,2008,23(1):41-42.
[5]馮麗華,袁飛鋒,石秋杰.高效液相色譜法測定金丹前列片中淫羊藿苷的含量[J].時珍國醫國藥,2008,19(3):690-691.
