6例強直性肌營養不良患者臨床與病理分析
2011-08-11 08:25:34張文娟李秋香畢方方孫新剛尹小玲梁靜慧
中風與神經疾病雜志 2011年12期
關鍵詞:癥狀
張 寧, 張文娟, 肖 波, 李秋香, 楊 歡, 畢方方, 孫新剛, 尹小玲, 梁靜慧
強直性肌營養不良(Myotonic dystrophy,DM),是一種累及全身多系統的常染色體顯性遺傳疾病[1],男性多于女性,主要臨床表現為肌強直、肌無力、肌萎縮,常伴有禿發、白內障、智力障礙、心律失常、糖尿病、周圍神經受累等。本研究總結分析中南大學湘雅醫院神經內科自2009年9月~2010年10月6例強直性肌營養不良患者資料,以期加強臨床醫生對該病的認識,進一步探討其發病機制。
1 資料與方法
1.1 一般資料 6例患者均為男性,年齡8~43歲,平均26.2歲;發病年齡3~37歲,平均年齡14.5歲;病程4~40年,平均12年。僅有1例患者其爺爺奶奶為姑舅親結婚。
1.2 臨床表現 6例患者均慢性起病,有不同程度的肌緊張,肌強直,用筷、書寫均不靈活,精細動作差,行走緩慢,靜息時全身發緊,寒冷時加重,反復活動后緩解。其中1例以雙手肌肉發緊、強直為首發癥狀,手握拳后松開困難,余5例均以雙下肢肌強直首發,起步費力。4例患者叩擊可見肌球征;僅1例有肌無力、肌萎縮,2例有脫發,1例伴智力障礙、吞咽困難、講話不清,2例有張口費力。
1.3 實驗室資料 6例患者血清肌酸激酶(CK)4例正常,2例輕度增高(CK 478.1 ~532.6U/L);肌電圖均示肌源性損害,可見強直電位發放。
1.4 方法 6例患者均在局麻下行左肱二頭肌開放式骨骼肌活體組織檢查,取材標本經液氮-異戊烷快速冷凍,7μm冰凍連續切片,行蘇木素-伊紅(HE)染色、Gomori染色、PAS染色和油紅(ORO)染色等特殊染色、還原型輔酶I四氮唑還原酶(NADHTR)染色、琥珀酸脫氫酶(SDH)染色、腺苷三磷酸環化酶(ATPase)、細胞色素C氧化酶(COX)、酸性磷酸酶染色、單磷酸腺苷脫氨酶(AMP)染色等酶組織化學染色,光鏡下病理分析。
2 結果
光鏡下病理改變:6例HE染色均可見:肌纖維大小不等,不同程度的肌纖維萎縮,1例可見角形纖維(見圖1);5例可見不同程度的肌纖維變性壞死;6例未見明顯再生肌纖維,1例可見肌分裂慢性病理改變;3例可見核內移、核聚集(見圖2),1例有鏈狀核(見圖3);1例結締組織重度增生,5例為輕度增生;間質均可見少量炎性細胞浸潤;Gomori染色均未見破碎紅纖維(RRF);PAS染色陰性;2例ORO染色可見部分脂肪代謝異常,余4例未見明顯異常;AMP染色酶活性正常;NADH、SDH、COX可見酶活性局限性增高或減低,肌原纖維網紊亂,呈蟲噬樣改變(見圖4);4例酸性磷酸酶染色酶活性增高;2例可見ATPase染色有肌纖維群組化,1例為I型肌纖維優勢(見圖5),另1例II型肌纖維群組化(見圖6)。
3 討論
DM是一種常染色體顯性遺傳異質性疾病,是成人最常見的一種肌營養不良,不僅僅有骨骼肌癥狀,還出現中樞神經系統癥狀(智力、情感障礙)、脫發、心臟傳導速度異常、眼部癥狀(白內障、視網膜色素變性等)、內分泌異常(腎上腺、胰島素分泌異常、甲狀腺功能異常等)、周圍神經受損,亦稱萎縮性肌強直。
DM病因和發病機制的研究經歷了長期的過程。目前認為其發病與離子通道有關[2]。正常肌細胞膜去極化引起鈉離子通道短暫快速的開放,大量Na+內流,同時氯離子通道緩慢打開,Cl-進入細胞內使膜電位回到靜息狀態。鈉離子通道突變引起Na+反復進入,重復引起去極化,如:高鉀性周期性癱瘓、低鉀性周期性癱瘓、波動性肌強直、惡性高熱等。Cl-通道突變使Cl-進入減少,復極化減少,而去極化相對延長,肌肉反復放電從而引起肌強直[3,4],如:先天性肌強直、強直性肌營養不良等。Mankodi A[5,6]等學者先后在轉基因動物模型和強直性肌營養不良患者中報道,CUG或CCUG重復擴增,其轉錄產物mRNA在肌組織中高表達,使跨膜Cl-通道水平減少,從而引起肌強直。
按基因突變位點和臨床表現DM分為DM1和DM2兩型,最近研究又發現 DM3型。1992年,Brook,Buxton等國外學者已確定DM1系第19號染色體長臂上(19q13.3)的DMPK基因突變,3’-端非翻譯區的(CTG)n三核苷酸重復序列擴增,編碼萎縮性肌強直蛋白激酶(dystrophia myotonic protein kinase,DMPK),然而這種酶的作用尚不明確,可能是在鈣離子平衡穩態、信號轉導以及骨骼肌纖維變性方面起作用。正常人該基因位點有4~37個CTG的重復序列,而DM患者的重復序列n可擴增至50~2000或更多至4000(見表1),其擴增程度與發病年齡及臨床癥狀相關,重復單元越多,臨床癥狀越嚴重。目前該蛋白酶致病動物模型已建立[7],最近國內趙曉萍[8]等人發現一個非CTG非CCTG重復擴展型強直性肌營養不良家系,該研究表明除CTG、CCTG短串聯重復序列異常擴增外,可能存在新的DM致病基因。DM2型系 3號染色體長臂上(3q21.3)鋅指蛋白9基因(ZNF9基因)的CCTG四核苷酸重復序列倍增突變引起[9],該基因編碼鋅指結構蛋白。正常情況下ZNF9基因位點有20~27個CCTG重復序列,DM2型患者可擴增達2000以上(見表1)。國外 Isabelle Le Ber[10]等發現一個新的致病基因突變位點,位于15號染色體長臂(15q21-24)上,該基因引起的肌病稱為強直性肌營養不良DM3型。發病機制尚不十分明確。DM患者若母親系基因攜帶者,后代具有發病提前和病情加重的遺傳早現現象[11],因此,胎兒產前診斷意義突顯重要。
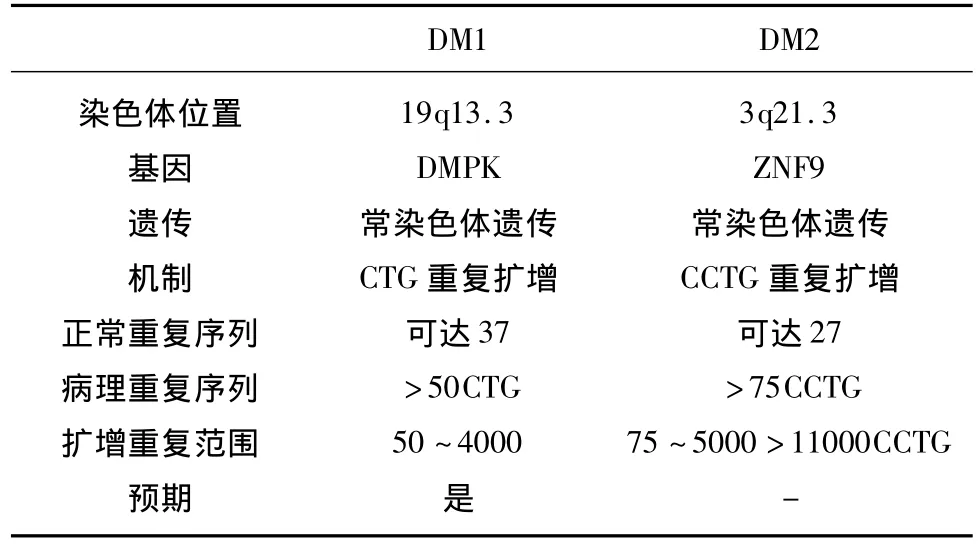
表1 DM1和DM2的病因
DM臨床癥狀多種多樣,DM1與DM2臨床表現相似,共同的癥狀有肌強直、肌無力、肌萎縮、白內障、心臟傳導改變、智力障礙、嗜睡、胰島素抵抗、睪丸萎縮、肌痛、禿頂等[12]。本研究總結分析的6例患者臨床表現除肌強直外,僅1例有肌無力、肌萎縮,余5例查體未見明顯肌力低下及肌萎縮,患者主訴肌無力時可能為肌緊張、肌強直導致活動不靈活,精細動作不佳,并非真正無力。2例有脫發、禿頭,無血糖升高、性激素等內分泌異常的表現。2例有張口費力,無吞咽困難、飲水嗆咳,該患者張口費力可能為開始肌緊張,啟動困難,反復張口活動后可緩解。1例其爺爺奶奶為姑舅親結婚。無眼睛、心臟及消化道癥狀。DM1按發病年齡和疾病進展程度分3型:(1)經典型;(2)先天型;(3)輕型。經典型患者一般首發癥狀是遠端肢體肌強直,本研究中有1例患者以雙手肌肉發緊,強直為首發癥狀,符合這一經典臨床特點。DM手部小肌肉萎縮有時出現較早,此時容易誤診,應注意與運動神經元病中的進行性脊肌萎縮癥(SMA)、肌萎縮側索硬化(ALS)、平山病、伴邊緣空泡的遠端型肌病等相鑒別(見表2)。部分DM患者可能會出現吞咽困難、構音障礙等[13],本研究中有1例伴吞咽困難、講話含糊的患者;DM2早期表現近端肌受累,又稱為近端型強直性肌營養不良,面肌較少累及,病變無特異性[12]。DM3型除有典型臨床癥狀外,還伴有額顳葉癡呆[10]。本研究中1例有智力障礙的患者,其理解力、計算力等均差,此時應仔細詢問病史,認真查體,結合其他臨床特征及輔助檢查以便做出診斷。DM晚期肌萎縮可累及近端,如累及面肌、眼外肌,導致面部表情減少,上瞼下垂,頰部消瘦,導致“斧形臉”,是本病的特征性面容,本研究中6例患者均無此特征性表現。

表2 DM、青少年型SMA、ALS、平山病、伴有鑲邊空泡的遠端型肌病比較
國內崔毅[14]等曾報道肌電圖隨病理特征不同,出現明顯不同的電位,DM肌纖維病變早期,EMG表現自發電位、短棘波多相電位、復合性電位、神經傳導速度(NCV)正常;中期,EMG可見肌強直電位、失神經復合型電位、群多相電位、短棘波多相電位和再生電位;晚期,EMG出現強直性電位、自發電位、復合性電位、失神經電位,呈神經性損害、肌源性損害,NCV均有減慢,部分未能引出神經動作電位,部分出現神經傳導阻滯(包括MCV、SCV)。本研究中6例DM患者肌電圖電生理檢查均提示肌源性損害,有強直電位發放,可排除神經源性疾病,僅1例NCV減慢,余5例NCV在正常范圍。血清肌酸激酶(CK)檢測,4例患者CK正常,2例輕度增高(CK 478.1~532.6U/L)。
6例DM患者肌肉標本組織學及酶學病理變化,HE染色均可見肌纖維大小不等,不同程度的肌纖維萎縮,1例可見角形纖維(見圖1),與神經源性病理改變的小角化肌纖維難鑒別,應緊密結合臨床分析。6例均未見明顯再生肌纖維,1例可見肌纖維分裂,肌分裂是代表肌原纖維的芽生,為慢性神經肌肉疾病的表現之一;3例可見典型核內移、核聚集(見圖2),1例有鏈狀核(見圖3),均為DM病理的特征性變化,核聚集、核內移與鏈狀核同時存在時,首先考慮DM這一診斷的可能性;2例ORO染色可見部分脂肪代謝異常,脂肪代謝包括脂肪酸的活化、轉運和氧化等過程,其中多個環節異常均可造成脂肪酸代謝障礙,需緊密結合臨床判斷其是否為脂質沉積性肌病;NADH、SDH、CCO可見酶活性局限性增高或減低,肌原纖維網紊亂,呈蟲蝕樣改變(見圖4),肌原纖維網紊亂、蟲蝕樣改變為細胞內酶的慢性改變;ATPase染色可見2例群組化肌纖維,1例可見選擇性I型肌纖維萎縮及I型肌纖維占優勢(見圖5),另1例可見II型肌纖維群組化(見圖6),余4例任意視野下均可見兩型肌纖維呈鑲嵌式分布。DM發病初期,I型與II型呈馬賽克分布,隨著疾病的進展,失神經支配,I型肌纖維逐漸占優勢。DM病理變化的輕重與肌強直癥狀無明顯相關性,與肌力低下的程度有關。DM的肌肉病理與臨床表現相關,臨床表現愈重肌肉損害愈明顯[15]。分析本研究6例患者臨床表現及病理結果,與該文獻結果一致。
近年來由于分子生物學技術的發展,DM等遺傳性肌肉疾病病理診斷已進入基因水平,基因檢測為DM診斷的基本手段,但對經濟條件要求很高,大部分醫療機構難普及,骨骼肌活檢病理分析對此病的診斷有一定價值。
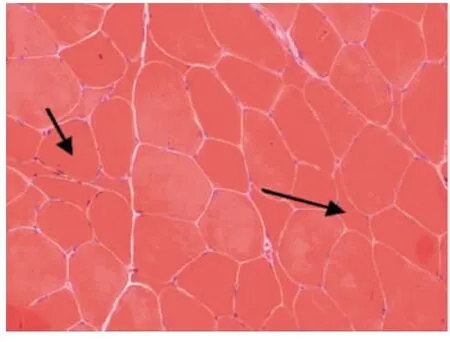
圖1 肌纖維大小不等,箭頭處為小角化肌纖維 HE×200
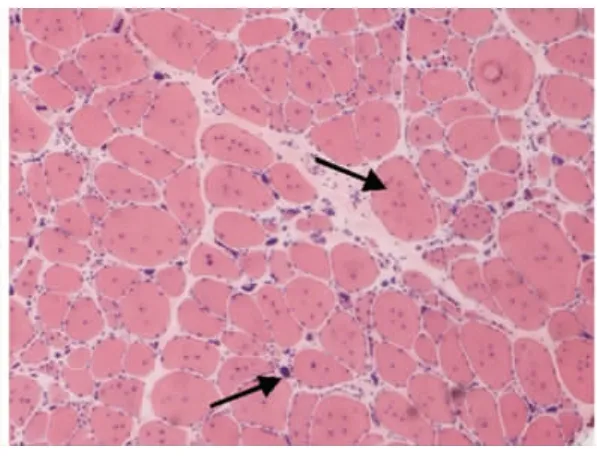
圖2 箭頭處為核內移,核聚集HE×100
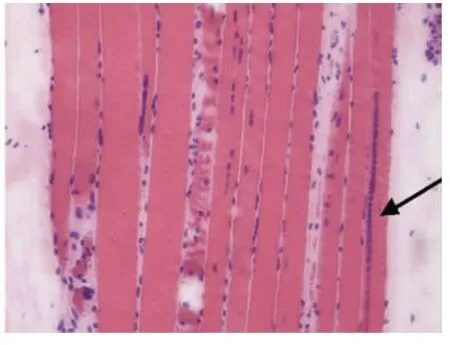
圖3 箭頭處為核聚集,鏈狀核HE×200
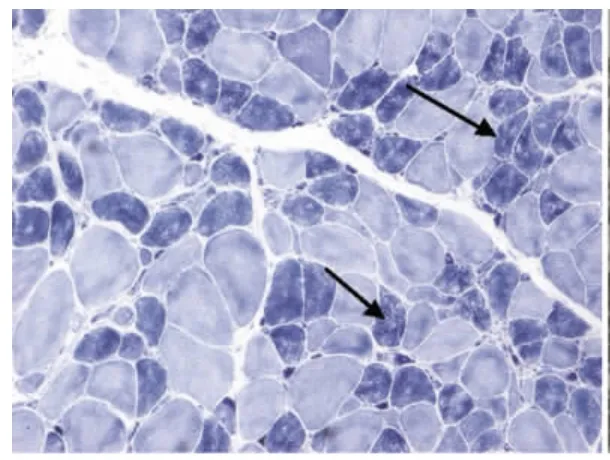
圖4 肌原纖維網紊亂,蟲噬狀肌纖維NADH×100
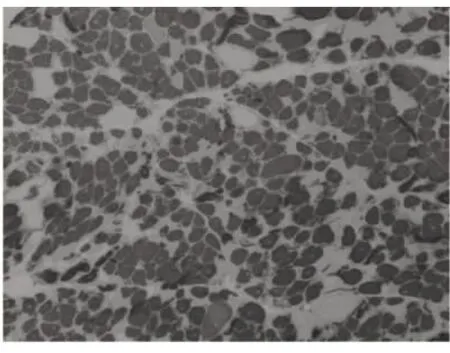
圖5 Ⅰ型肌纖維群組化ATP4.3×100

圖6 Ⅱ型肌纖維群組化ATP4.3×10
[1]Zerylnick C,Torroni A,Sherman SL,et al.Normal variation at the myotonic dystrophy locus in global human populations[J].Am J Hum Genet,1995,56(1):123 -130.
[2]Ranum LW,Day JW.Myotonic dystrophy:RNA pathogenesis comes into focus[J].Am J Hum Genet,2004,74(5):793 - 804.
[3]楊華艷,陳宜張.骨骼肌-神經系統離子通道病[J].基礎醫學與臨床,2005,25(11):992 -998.
[4]江新梅,胡 靜.離子通道與骨骼肌疾病[J].中國實用內科雜志,2007,27(6):876 -878.
[5]Mankodi A,Logigian E,Callahan L,et al.Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat[J].Science,2000,289(5485):1769-1773.
[6]Mankodi A,Takahashi MP,Jiang H,et al.Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscal in myotonic dystrophy[J].Mol Cell,2002,10(1):35 -44.
[7]Orengo JP,Chambon P,Metzger D,et al.Expanded CTG repeats within the DMPK 3’UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy[J].Proc Natl Acad Sci USA,2008,105(7):2646 -2651.
[8]趙曉萍,謝惠君,鄭惠民,等.一個非CTG非CCTG重復擴展型強直性肌營養不良家系[J].中華醫學遺傳學雜志,2004,21(5):459 -462.
[9]Christina LL,Kenneth R,Melinda LM,et al.Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9 [J].Science,2001,293(5531):864 -867.
[10]Le Ber I,Martinez M,Campion D,et al.A non-DM1,non-DM2 multisystem myotonic disorder with frontotemporal dementia:phenotype and suggestive mapping of the DM3 locus to chromosome 15q21-24[J].Brain,2004,127:1979 -1992.
[11]蘇敬敬,王桂斌,趙武偉,等.CTG重復數與強直性肌營養不良的遺傳早現現象[J].中風與神經疾病雜志,2003,20(4):337 -339.
[12]Schoser B,Timchenko L.Myotonic dystrophies 1 and 2:complex diseases with complex mechanisms[J].Curr Genom,2010(11):77 -90.
[13]De Swart BJM,Van Engelen BGM,Vande Kerkhof JPBM,et al.Myotonia and flaccid dysarthria inpatients with adult onset myotonic dystrophy[J].J Neurol Neurosurg Psychiatry,2004,75:1480 -1482.
[14]崔 毅,張永巍,王 曄,等.強直性肌營養不良的臨床、肌肉病理與肌電圖神經傳導速度對比研究[J].臨床神經電生理雜志,2002,11(2):79 -81.
[15]王 曄,鄭惠民,謝惠君,等.強直性肌營養不良的臨床與肌肉病理研究[J].中風與神經疾病雜志,2000,17(5):297 -298.
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
