鏈接基團對固載手性Salen Mn(Ⅱ)催化不對稱環氧化反應的影響
2011-11-09 08:06:12羅云飛傅相鍇鄒曉川王長煒胡小艷賈紫永張懷志
無機化學學報 2011年7期
關鍵詞:催化劑
羅云飛 傅相鍇*, 鄒曉川,2 王長煒 胡小艷 賈紫永 張懷志
(1西南大學化學化工學院,應用化學研究所,重慶市應用化學重點實驗室,三峽庫區生態環境教育部重點實驗室,重慶 400715)
(2重慶教育學院生物化學工程系,重慶 400067)
鏈接基團對固載手性Salen Mn(Ⅱ)催化不對稱環氧化反應的影響
羅云飛1傅相鍇*,1鄒曉川1,2王長煒1胡小艷1賈紫永1張懷志1
(1西南大學化學化工學院,應用化學研究所,重慶市應用化學重點實驗室,三峽庫區生態環境教育部重點實驗室,重慶 400715)
(2重慶教育學院生物化學工程系,重慶 400067)
制備了二胺和二酚修飾的晶態有機聚合物-無機雜化載體低聚苯乙烯基膦酸-膦酸氫鋯(LCZSPP)軸向固載手性Salen Mn(Ⅱ)催化劑,將其應用于非官能烯烴的多相不對稱環氧化反應。研究了兩類軸向連接基團及助催化劑在催化不對稱環氧化反應中的影響。結果表明,通過二胺為鏈接基團固載的催化劑在加入軸向助劑(NMO)的情況下,轉化率和對映選擇性都有較大提高。然而,與文獻報道相反,二酚固載的催化劑在不添加軸向助劑的情況下,轉化率和對映選擇性都比添加軸向助劑有明顯提高。此外,制備的固載催化劑可以很容易從反應體系中分離出來,并且在重復使用10次以上后,催化活性和對映選擇性都沒有明顯的降低。
低聚苯乙烯基膦酸-膦酸氫鋯;手性Salen Mn(Ⅱ);多相催化劑;非官能化烯烴的不對稱環氧化反應;軸向助催化劑
0 引 言
具有光學活性的環氧化物是合成許多天然產物、光學活性材料、光學活性藥物等重要有機合成中間體。現已成為當今研究的熱門課題之一。手性環氧化物是合成純的對映體分子的重要模塊,可以通過選擇性開環和官能團轉化方便地合成許多醫藥和精細化學品的重要中間體,特別是在合成藥物、生物活性化合物中有重要的應用[1-2]。目前制備手性環氧化物最重要的方法是烯烴的催化環氧化,而常用的催化劑為過渡金屬卟啉、酞菁以及Salen等的配合物[3-4]。人們發現手性Salen Mn(Ⅱ)配合物是非官能化烯烴不對稱環氧化反應最有效的催化劑[5-6],然而,均相催化劑存在著難以分離,容易形成Salen Mn(Ⅱ)的μ-oxo-Mn(Ⅱ)無催化活性二聚體,造成其不能循環使用,限制了它的廣泛應用[7]。為解決這一問題,目前,已成功地將此類配合物負載到無機、有機或者有機-無機復合載體上,并且部分地實現了催化劑的循環使用[8~10]。更值得指出的是,在大多數情況下,固載型Salen Mn(Ⅱ)催化劑均需要在昂貴的助催化劑參與下才能對非功能化烯烴的不對稱環氧化起到促進作用[11-13],這就使其在工業化應用中失去了意義。因此,研究助催化劑在不對稱環氧化反應中的作用機理,以及設計一類在沒有昂貴助催化劑參與下使環氧化反應順利進行的多相催化劑具有非常重要的工業意義。
近年來,本課題組探索合成了有機聚合物-無機雜化磷酸鋯新型催化劑載體系列ZSPP、ZPS-iPPA和ZPS-PVPA,采用側鏈接枝或軸向配位固載技術合成了這些載體固載的手性Salen Mn(Ⅱ)多相催化劑,在催化烯烴的不對稱環氧化反應中顯示了優良的催化活性、對映選擇性和卓越的重復使用性[14-22]。但ZSPP、ZPS-iPPA和ZPS-PVPA聚苯乙烯基膦酸-磷酸氫鋯系列載體均為無定型。本工作探索合成了晶態低聚苯乙烯基膦酸-磷酸氫鋯復合載體,并分別用有機二胺和二酚作為連接手臂,與手性Salen Mn(Ⅱ)進行軸向配位固載。詳細考察了兩種不同的連接手臂及助催化劑對非功能化烯烴不對稱環氧化反應催化性能的影響。
1 實驗部分
1.1 儀器和試劑
PK-6000型FTIR紅外光譜儀;DR UV-2550型紫外可見分光光度計;TAS-986G型原子吸收分光光度計;KYKY-EM3200型掃描電子顯微鏡;美國vecco公司原子顯微鏡(AFM);采用日本島津公司GC-2014型氣相色譜儀對反應進行測定。通過與標準產物比較計算產率及對映選擇性(ee)值。氫火焰離子化檢測器,N2為載氣。色譜柱為手性毛細管(HP19091G-B233,30 m×25 μm×0.25 μm),采用程序升溫程序,其中α-甲基苯乙烯(柱箱溫度:80℃,汽化溫度:230℃,檢測器溫度:250℃,載氣流速:34.0 mL·min-1,分流比:30.0)茚(柱箱溫度:80℃,汽化溫度:230℃,檢測器溫度:230℃,載氣流速:24.0 mL·min-1,分流比:30.0)
壬烷、N-甲基嗎啉氮氧化物 (NMO)、(1R,2R)-環己二胺(99%)、間氯過氧苯甲酸(m-CPBA)、α-甲基苯乙烯、茚均購自Alfa Aesar。其他試劑和溶劑均為市售分析純。
1.2 載體的制備
低聚苯乙烯基亞膦酸,低聚苯乙烯基膦酸的制備參見文獻[18]。
晶態層狀低聚苯乙烯基膦酸-磷酸氫鋯的制備是在聚四氟乙烯塑料杯中,將OSPNA(5.2 g,13 mmol)和NaH2PO4·2H2O(4.2 g,27 mmol)和50 mL水的混合液,在攪拌下滴加到 ZrOCl2·8H2O(6.4 g,20 mmol)和10 mL 40%HF(0.20 mol)的100 mL溶液中,75℃攪拌5 d,直到白色沉淀析出,離心過濾,用水洗滌至濾液為pH=5,甲醇洗滌(40 mL×3),70℃真空干燥24 h。產率約70%。IR(KBr,cm-1),3027,2 925(CH),2370(O=P-OH),1603,1494,1453,754 (-C6H5),1 267(P=O),1 067,1 013)。
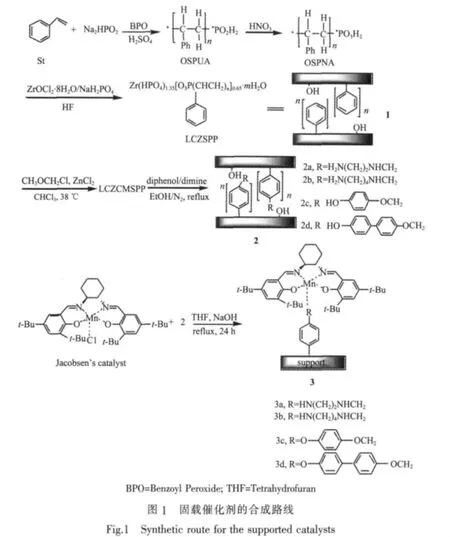
氯甲基化晶態低聚苯乙烯基膦酸-磷酸氫鋯、胺化和酚羥基化載體的制備參照文獻[18-21]。結構如圖1中所示。
1.3 催化劑的制備
均相手性Salen Mn(Ⅱ)的制備參照文獻[23]。分別將0.5 g胺化和酚羥基化載體2a、2b、2c和2d,均相手性Salen Mn(Ⅱ)催化劑 (2.5 g),適量氫氧化鈉,40 mL四氫呋喃,加入到100 mL三頸瓶中回流反應12 h。過濾,分別用甲苯、二氯甲烷及去離子水洗滌濾餅至洗液經原子吸收檢測不到錳元素存在,真空干燥24 h得棕色粉末狀固載催化劑,分別記作3a、3b、3c和3d。催化劑3a IR(KBr cm-1):2930-2870(CH),1 615,1 495,1 455(-C6H5),1 624-1 627(C=N),520 (Mn-N)。DR UV-Vis:251,423 nm;催化劑 3b IR (KBr cm-1):2 950-2 880 (C-H),1 625,1 500,1 460 (-C6H5),1 624-1 635(C=N),521(Mn-N)。DR UV-Vis:253,425.5 nm。催化劑3c IR(KBr cm-1):2 935-2 850 (C-H),1 611,1 492,1 450(-C6H5),1 620-1 635(C=N),520(Mn-N)。DR UV-Vis:252,428.5 nm;催化劑 3d IR (KBr cm-1):2 945-2 850(C-H),1 615,1 02,1 460 (-C6H5),1 625-1 645(C=N),520(Mn-N)。DR UV-Vis:251,428.2 nm。
2 結果與討論
2.1 催化劑的微觀形貌
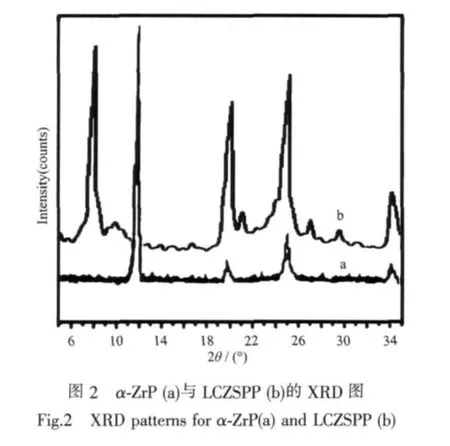
圖2為載體的粉末衍射圖,從圖中可知樣品在2θ=8.23°,19.48°,24.54°存在3個很強的吸收峰,對應的內層間距分別為1.065、0.443和0.336 nm。和α-ZrP相比,最大的不同是在2θ=8.23°出現了1個新的吸收峰,其層間距(1.065 nm)比α-ZrP的層間距(0.764 nm)大0.301 nm。表明,樣品的層間距為1.065 nm。層間距增大是由于樣品中存在大量剛性的苯環和無機基團所致。
固載催化劑3d的SEM、TEM及AFM照片如圖3、4、5。SEM和TEM圖顯示固載催化劑的顆粒的粒徑大小在幾十到100 nm范圍,結構疏松,擁有大量的納米通道、孔洞和空腔。從圖5a,5b可以看出固載催化劑中存在大量的孔洞,其大小為 13.0~15.2 nm。 因此催化劑可以為不對稱環氧化反應提供足夠的空間,這可能是固載化均相催化劑具有優良的催化活性和對映選擇性最主要的因素之一。


2.2 催化烯烴環氧化性能研究
以間氯過氧苯甲酸(m-CPBA)為氧化體系催化烯烴的不對稱環氧化反應,考察了反應時間、溫度、助催化劑等因素對催化結果的影響,并且與均相Jacobsen催化劑進行對比。

表1 m-CPBA體系中催化劑3a-3d催化烯烴不對稱環氧化反應[a] Table 1 Asymmetric epoxidation of alkenes catalyzed by 3a-3d with m-CPBA as oxidants[a]
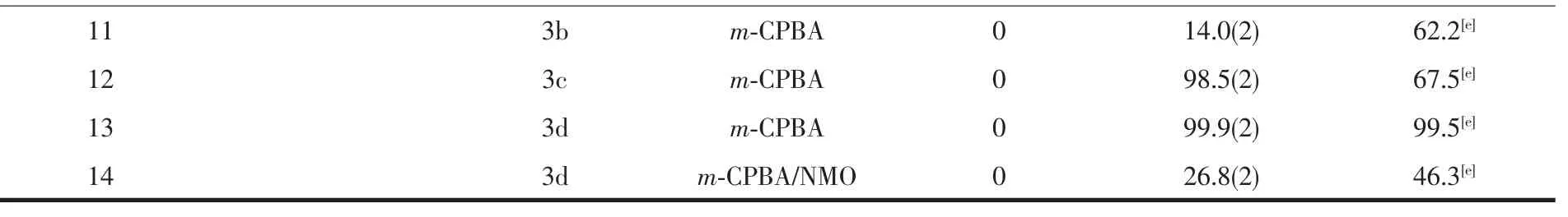
續表1
從表1中可以看出固載催化劑3a~3d表現出了與均相Jacobsen催化劑相近甚至更高的對映選擇性。尤其對于α-甲基苯乙烯,對映選擇性從53.5%增加到73.8%,對映選擇性的增加主要歸因于軸向連接基團與載體(LCZSPP)所組成的微環境以及載體納米孔道的限閾效應。
2.3 助催化劑效應
大量的文獻報道軸向添加劑對催化劑的活性和對映選擇性起到促進作用。本文中,對于有機二胺固載的催化劑(3a~3b),當加入助催化劑NMO,轉化率和對映選擇性都有較大的提高,這與文獻報道相一致。當加入助催化劑NMO,尤其對于α-甲基苯乙烯轉化率從42.5%增加到98.5%(表1),對映選擇性24.8%增加到73.1%。但是對于有機二酚手柄固載的催化劑(3c,3d)卻出現了截然相反的結果,即不加入比加入助催化劑,轉化率和對映選擇性不但沒有降低,反而大大提高,這與絕大多數先前報道的文獻相矛盾。尤其對于 α-甲基苯乙烯 (轉化率從 99.9%降低到32.8%;ee:從73.8%降低到14.9%;entries 6 and 7),相似的結果對于茚也同樣可以得到(entries 13 and 14)。原因可能是Mn-O化學鍵比Mn-N化學鍵具有更多離子性;另一個原因可能是酚氧基比脂肪族胺基具有更大的剛性和空間位阻。最近List小組[24]采用大位阻的手性聯二萘酚剛性配體的Salen Mn(Ⅱ)催化色素烯及其衍生物的不對稱環氧化反應有相似實驗現象的報道,但未給出具體的實驗數據。
實際上,作為連接手臂的酚氧基團具有和助催化劑NMO具有相似的電子結構和配位性質。不存在NMO的情況,催化劑3c與3d已經表現出來相當滿意的效果(entries 5 and 6,entries 12 and 13),因為在這里酚氧基團扮演了軸向連接基團和軸向助劑例如(NMO)的雙重作用。此時加入NMO后,NMO可能干擾不對稱環氧化反應中已經形成的較好的中間體或者過渡態的幾何構型與構象,從而影響手性誘導和催化效果。對于這些固載催化劑表現出反常現象的機理的研究正在進一步進行中。
2.4 不同溶劑與溫度的影響
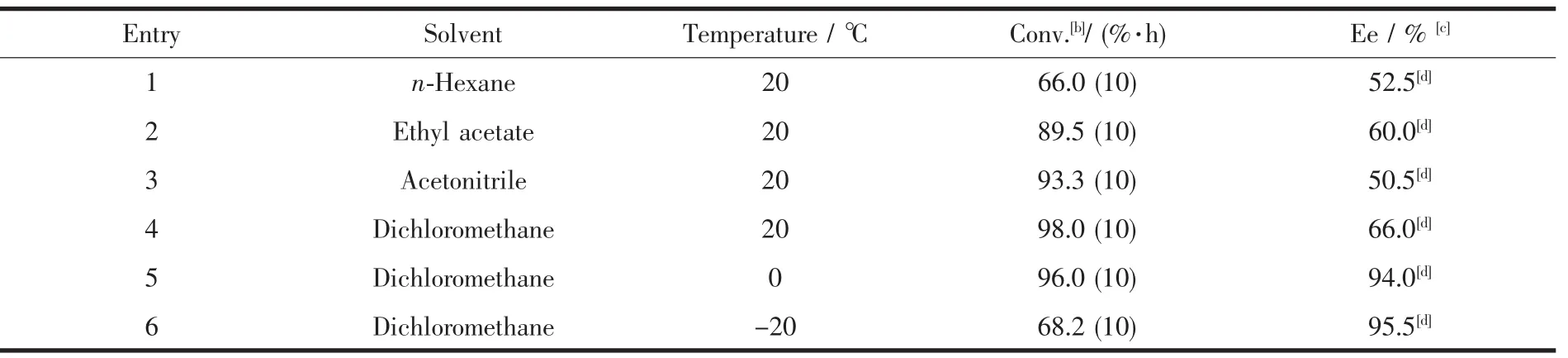
表2 固載催化劑3d在不同的溶劑和溫度條件下催化茚的環氧化反應[a]Table 2 Epoxidation of indene catalyzed by immobiled catalyst 3d in different solvents and temperature[a]
表2中概括了催化劑3d在不同溶劑和溫度條件下催化茚的不對稱環氧化結果。從表中可以看出,當用二氯甲烷做溶劑時,不論是轉化率還是對映選擇性都是最好的。同時,我們發現隨著溫度的降低,對映選擇性在增加,而轉化率略微降低。可能的原因是隨著溫度的降低,底物分子的有效碰撞減少,因此轉化率有所降低。同時可能隨著溫度的降低,更有利于環氧化產物中C-O鍵的形成和抑制過渡態的反轉造成更高的對映選擇性。
2.5 催化劑的循環使用
表3為固載化均相催化劑對α-甲基苯乙烯的循環使用情況。從表3中可以看出,和均相Jacobsen催化劑相比,固載催化劑3d在循環使用10次后仍然具有較高的催化活性。這主要歸因于載體的結構是由疏水的聚苯乙烯片段和具有親水性的以及具有納米層狀自組裝結構的無機磷酸鋯部分以及構成載體LCZSPP中的有機聚苯乙烯部分相互重疊、扭曲、平行、交叉、甚至纏繞以及層狀磷酸鋯部分形成了不同分子尺寸大小的孔徑、空洞、以及二級通道一起構成的微環境,使得固載催化劑表現出如此優異的循環性能。
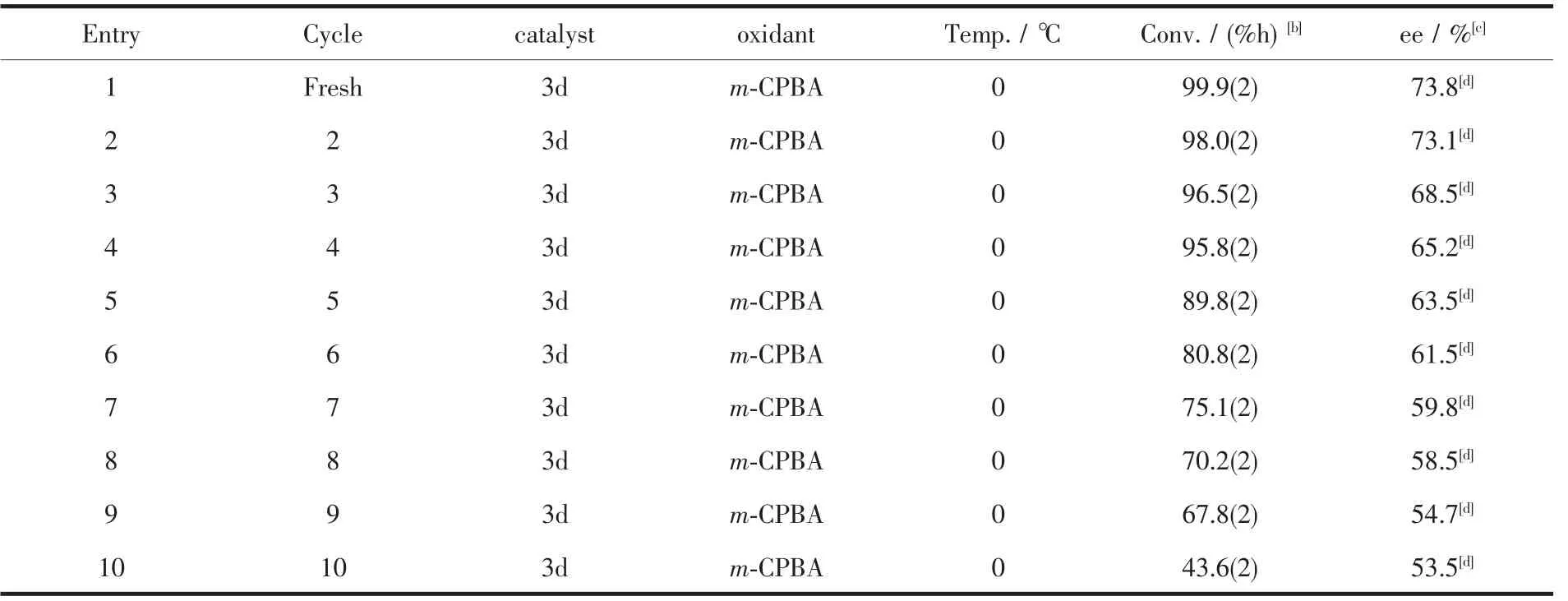
表3 固載催化劑3d催化α-甲基苯乙烯環氧化反應的循環使用性能[a]Table 3 Recycling of immobilized catalyst in the asymmetric epoxidation of α-methylstyrene with m-CPBA as the oxidant[a]
3 結 論
(1)用氫氟酸絡合法合成出了新型層狀晶態有機聚合物無機雜化低聚苯乙烯基膦酸-磷酸氫鋯載體材料。并將其用于固載手性Salen Mn(Ⅱ)催化不對稱環氧化反應中。
(2)制備的固載催化劑表現出了和均相催化劑相當或更高的催化活性,并且具有較好的重復使用性。
(3)對兩類不同鏈接手臂進行研究發現,二酚類鏈接手臂固載的手性Salen Mn(Ⅱ)催化劑在不加入昂貴的助催化劑情況下,就表現出了很好的對映選擇性和催化活性。
[1]Schurig V,Betschiner F.Chem.Rev.,1992,92:873-888
[2]Rukhsana I K,Noor-ul H K,Sayed H R A,et al.J.Catal., 2004,224:229-235
[3]GUO Feng(郭鋒),ZHANG Hong-Qi(張洪起),FENG Lei (馮磊),et al.Chem.J.Chinese Universities(Gaodeng Huaxue Xuebao),2008,22:972-977
[4]ZHAO Dong-Min(趙東敏),ZHAO Ji-Quan(趙繼全),ZHAO Shan-Shan(趙姍姍).Chinese Sci.Bull.(Kexue Tongbao), 2007,52:886-892
[5]Zhang W,Jacobsen E N.J.Org.Chem.,1991,56:2296-2298
[6]Katsuki T.Adv.Synth.Catal.,2002,344:131-147
[7]Jacobsen E N,Deng L,Furukawa Y.Tetrahedron,1994,50: 4323-4334
[8]McGarrigle E M,Gilheany D G.Chem.Rev.,2005,105:1563-1602
[9]Baleizo C,Garcia H.Chem.Rev.,2006,106:3987-4043
[10]Fraile J M,Garci J I,Mayoral J A.Chem.Rev.,2009,109: 360-417
[11]Palucki M,Pospisil P J,Zhang W.J.Am.Chem.Soc.,1994, 116:9333-9334
[12]Palucki M,Mccormick G J,Jacobsen E N.Tetrahedron Lett., 1995,36:5457-5460
[13]Wang D P,Wang M,Wang X N.J.Catal.,2006,237:248-254
[14]Ma X B,Fu X K.J.Mol.Catal.A:Chem.,2003,195:47-53
[15]Sui Y,Fu X K,Zeng R Q,et al.J.Mol.Catal.A:Chem., 2004,217:133-138
[16]Wu X J,Ma X B,Ji Y L,et al.J.Mol.Catal.A.Chem., 2007,265:316-322
[17]BAO He-Bin(包河彬),FU Xiang-Kai(傅相鍇),BAI Ruo-Fei (白若飛),et al.Chem.J.Chinese Universities(GaodengHuaxue Xuebao),2008,25:927-931
[18]Bai R F,Fu X K,Bao H B,et al.Catal.Comm.,2008,9: 1588-1594
[19]TU X B,Fu X K,Hu X Y,et al.Inorg.Chem.Commun.,2010, 13:404-407
[20]Gong B W,Fu X K,Chen J X,et al.J.Catal.,2009,262:9-17
[21]Ren W S,Fu X K,Bao H B,et al.Catal.Commun.,2009, 10:788-793
[22]Zou X C,Fu X K,Li Y D,et al.Adv.Synth.Catal.,2010, 352:163-170
[23]Zhang W,Loebach J L,Wilson S R,et al.J.Am.Chem. Soc.,1990,112:2801-2803
[24]Saihu L,Benjamin L.Angew.Chem.Int.Ed.,2010,49:628-631
[25]Zhang H D,Xiang S,Xiao J L,et al.J.Mol.Catal.A,2005, 238:175-184
[26]Kureshy R I,Ahamd I,Khan N H,et al.J.Catal.,2005, 235:28-34
Effect of Axial Linker Group on Heterogeneous Chiral Salen Mn(Ⅱ)Catalyzed Enantioselective Epoxidation of Unfunctionalized Olefins
LUO Yun-Fei1FU Xiang-Kai*,1ZOU Xiao-Chuan1,2WANG Chang-Wei1HU Xiao-Yan1JIA Zi-Yong1ZHANG Huai-Zhi1
(1College of Chemistry and Chemical Engineering,Research Institute of Applied Chemistry Southwest University, The Key Laboratory of Applied Chemistry of Chongqing Municipality,The Key Laboratory of Eco-environments in Three Gorges Reservoir Region(Ministry of Education),Chongqing 400715,China)
(2Department of Biological&Chemical Engineering,Chongqing Education College,Chongqing 400067,China)
The chiral Salen Mn(Ⅱ)was axially immobilized onto a diamine or diphenoxyl linker modified layered crystalline hybrid material-zirconium oligostyrenylphosphonate-phosphate (LCZSPP)synthesized in this lab.The influence of the two types of linkers and axial additives on the catalytic performance of the heterogeneous Salen Mn(Ⅱ)was investigated.The results demonstrate that the conversion and enantioselectivity(ee)value increase in the presence of axial additive such as NMO for the Salen Mn(Ⅱ) immobilized onto LCZSPP via diamine linkers while the opposite is true for the immobilized Salen Mn(Ⅱ) via diphenoxyl.In addition,the prepared catalysts could be conveniently separated from the reaction system by simple precipitation in hexane,and could be reused at least ten times without significant loss of activity and enantioselectivity.
zirconium oligostyrenylphosphonate-phosphate;chiral Salen Mn(Ⅱ);heterogeneous catalysts;enantioselective epoxidation of unfunctionalized olefins;axial additive
O614.3;TQ426.65
A
1001-4861(2011)07-1302-07
2011-01-15。收修改稿日期:2011-03-21。
國家科技部科技中小型企業技術創新基金(No.09C26215112399)和國家人力資源和社會保障部留學人員回國創業啟動支持計劃(人社廳發2009(143號))資助項目。
*通訊聯系人。E-mail:fxk@swu.edu.cn
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
