放射性核素在氧化物、磷酸鹽和黏土礦物表面吸附:熱力學和微觀結構認識
2012-01-04 09:03:44范橋輝牛智偉許君政郭治軍吳王鎖
核化學與放射化學 2012年3期
關鍵詞:界面
范橋輝,牛智偉,許君政,郭治軍,吳王鎖
蘭州大學 核科學與技術學院 放射化學與核環境研究所,甘肅 蘭州 730000
放射性核廢物的妥善處理和安全處置是制約我國核能發展戰略的關鍵因素之一,而放射性核素在環境介質中吸附行為的研究是放射性廢物處置過程中的基礎研究內容之一[1]。利用實驗測得的放射性核素在固相和液相間的分配系數以及其在固相中的擴散系數,可以比較準確的預測和評估放射性核素在環境介質中的遷移行為,為核廢物妥善處理和安全處置提供有效的理論指導。放射性核素在固液界面上的吸附機理主要包括表面配位作用、表面沉淀和離子交換反應等[2]。放射性核素在環境介質中的吸附行為受到多種因素控制或影響,如pH、離子強度、天然有機質、吸附質濃度和固體表面電荷性質、功能基團密度等,從而給人們對其吸附行為構建模型和探討吸附機理帶來了極大的困難。盡管大量研究工作對放射性核素在固液界面上的吸附機理進行了表觀和微觀上的探索和解釋,但這些實驗所得的模型和熱力學結論的正確性或合理性主要取決于實驗過程的合理與否。通過大量研究工作總結,發現通過以下7個主要步驟可得到相對準確、系統和科學的界面吸附信息:
(1)對吸附劑進行詳細的結構、形貌、拓撲等物理化學性質的表征,如XRD對吸附材料的結構表征,FTIR對吸附劑表面功能基團的表征, BET或ESA法測定固體顆粒的尺寸大小和比表面積等關鍵性質;
(2)吸附劑材料的表面電荷性質表征,吸附劑表面電荷密度與吸附體系pH之間的關系和吸附劑表面零電荷點是影響放射性核素吸附種態、過程和機理的重要參數;
(3)通過對固體材料的表面進行連續電位滴定,研究表面可變電荷位的質子化和去質子化反應的平衡常數;
(4)開展相關的靜態吸附研究,考察pH、離子強度、吸附質濃度和天然有機質等對放射性核素在固液界面吸附的影響;
(5)對放射性核素的表面吸附種態進行研究,考察放射性核素在固液界面上的微觀空間結構等信息;
(6)綜合以上結果得到宏觀和微觀結構,利用表面配位模型對放射性核素在固液界面上的吸附機理和過程進行探討;
(7)利用先進光譜技術對所得模型和機理進行分子或原子水平上的論證。
本文擬通過幾個具體實例分別對每一步驟進行詳細闡述。這些實例中,主要選取蘭州大學放射化學與核環境研究所針對放射性核素在固液界面吸附的研究成果,涉及放射性核素在氧化物(如氧化鋁、氧化鈦等)、磷酸鹽(ZrP2O7、Zr2O(PO4)2和Th4(PO4)4P2O7)、天然黏土礦物(膨潤土和凹凸棒石黏土)和石灰性土壤等上的吸附。吸附質主要涉及Cs(Ⅰ)、Ni(Ⅱ)、Eu(Ⅲ)、Th(Ⅳ)和U(Ⅵ)等。
1 固體表面材料的表征
1.1 固體材料的結構、拓撲性質表征
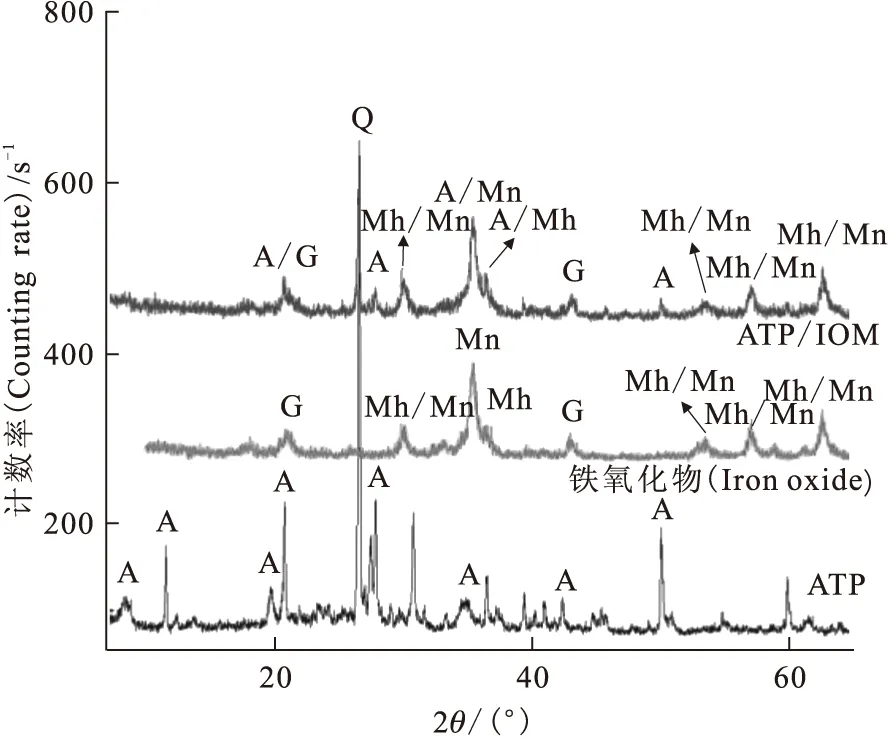
圖1 凹凸棒石黏土和鐵氧化物-凹凸棒石黏土磁性復合材料的XRD衍射圖
為了研究吸附質與吸附劑的相互作用機理,需對吸附材料的結構、形貌、表面功能基團等信息進行詳細的表征。通常XRD和SEM分別被用來表征吸附材料的基本結構和形貌。圖1和圖2分別為凹凸棒石黏土(ATP)和鐵氧化物-凹凸棒石黏土磁性復合材料(ATP/IOM)的XRD衍射圖和電子掃描電鏡形貌圖[3]。由圖1可知,鐵氧化物-凹凸棒石黏土磁性復合材料中除了2θ=20.7°、26.6°及42.5°處特征峰外,凹凸棒石其余特征峰均消失,說明凹凸棒石的結構可能因為鐵氧化物的負載導致凹凸棒石晶格所謂的混亂[4]。在復合材料中其他峰位置與標準圖譜JCPD中鐵氧化物XRD譜非常接近:Fe3O4磁鐵礦(89-3854,2θ=30.088°、35.439°、37.071°、43.07°、53.432°、56.958°和62.546°)或α-Fe2O3赤磁鐵礦(89-5892,2θ=30.266°、35.651°、37.294°、43.332°、53.766°、57.319°和62.949°)[5]。另外,2θ=35.42°和36.62°處的峰位置可能是由ATP/IOM復合材料造成的。由圖2可知,凹凸棒石呈纖維狀且一些纖維聚集形成棒狀,單個纖維直徑約40~60 nm,長0.5~1 μm。而鐵氧化物-凹凸棒石磁性黏土復合材料表面明顯變得更緊湊、粗糙,粒徑更小且邊角較平滑,這可能是由于鐵氧化物的引入使其形貌發生改變(圖2(a)—(c))所致。

圖2 凹凸棒石黏土和鐵氧化物-凹凸棒石黏土磁性復合材料的SEM
圖3為4種土壤樣品的紅外圖譜對比,分別為除碳酸鹽土壤(DCS)、Ca型石灰性土壤(CaCS)、吸附鈾酰離子Ca型石灰性土壤(U(Ⅵ)-CaCS)和吸附鈾酰離子除碳酸鹽土壤(U(Ⅵ)-DCS)。由圖3可知:3 620 cm-1和3 420 cm-1附近的峰分別對應AlO—H 和FeO—H的吸收峰;1 030 cm-1和467 cm-1處的峰為Si—O—Si振動,779 cm-1處的峰可能是由δ(O—Si)的伸縮振動引起;528 cm-1處的峰則對應O—P—O 的面外扭曲振動;而1 630 cm-1處的峰對應于水的彎曲振動[6-7]。對于Ca型石灰性土壤中在1 434 cm-1和875 cm-1處的吸收峰由CaCO3的官能團振動引起,但在除碳酸鹽土壤中已經消失,表明碳酸鹽組分被完全去除。CaCO3在1 434 cm-1和875 cm-1標準振動吸收在DCS、U(Ⅵ)-DCS和U(Ⅵ)-CaCS樣品中消失,說明石灰性土壤中CaCO3組分參與U(Ⅵ)在其表面反應。
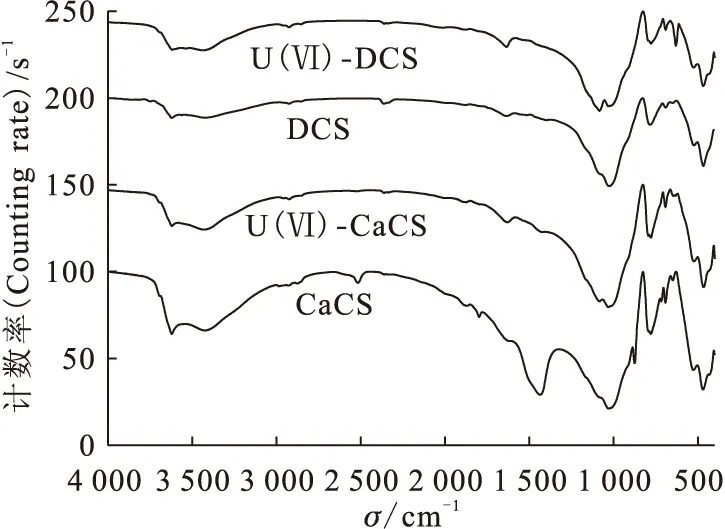
圖3 4種土壤樣品的紅外圖譜[8]
1.2 固體材料表面性質表征
1.2.1固體表面電荷產生 離子或有機質在固液界面的物理化學反應中,固體顆粒物的表面電荷和表面電勢是決定吸附質在其表面上的吸附過程、微觀吸附種態及吸附機理的重要參數。表面電荷/電勢的改變對吸附質在固液界面上的化合態、表面反應活化能等均有重要的影響,從而導致離子或有機質在固液界面上的吸附反應方向和吸附/解吸反應動力學等的改變。固體顆粒物的表面主要通過以下3種方式形成表面電荷[9],即固體表面上的化學反應、固相表面的晶格缺陷和晶格內的同晶置換以及表面吸附疏水性的化合物或者表面活性劑。
(1)固體表面上的化學反應
固體表面一般都帶有可離子化的官能團,如—OH、—NH2、—COOH、—OPO2H2、—SH等。可離子化官能團的質子轉移作用可致使固體表面產生電荷,表觀上表現出酸堿兩性(即可發生質子化和去質子化反應)。由表面官能團上質子轉移而產生的電荷,稱之為可變電荷或者凈質子電荷。可變電荷強烈依賴于體系pH值,在低pH值時固體表面主要以正電荷狀態存在;當在較高的pH值條件下則固體表面主要表現出負電荷形態;而在某個特定的pH值下,固體表面電荷可以為零(此處對應的pH為零電荷點,pHpzc)。對于其他的固體表面可發生類似的質子轉移反應,可寫成普遍通式(1)和(2):
(1)

(2)
但當固體表面與存在于溶液中的溶質離子結合時亦可產生表面電荷。例如以下反應:
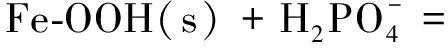
?Fe-OOPO3H-+H2O
(3)
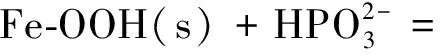
(4)
其中s表示固體[10]。
(2)固相的晶格缺陷和晶格內的同晶置換
固體晶格缺陷可使固體表面帶電荷。當晶格內SiO2四面體中的四價Si被三價Al取代,或Al2O3中鋁氧八面體中三價的Al被二價Mg、Fe等置換后,整個晶體帶一個單位的負電荷,如圖4所示。同晶置換作用是天然的黏土礦物骨架帶有電荷及擁有一定離子交換容量的主要原因。由結構缺陷或者同晶置換作用而產生的表面電荷幾乎不受固體所處溶液pH的影響,因此這類電荷被稱之為結構電荷或者永久性電荷。
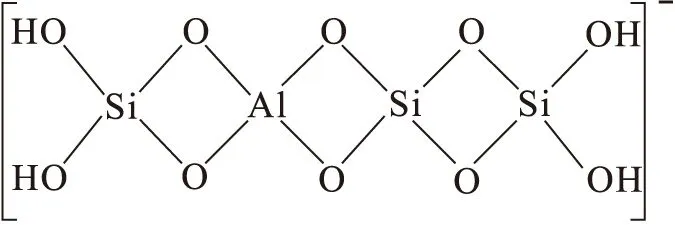
圖4 三價Al對四價Si的同晶置換作用
(3)表面吸附疏水性的化合物或者表面活性劑
通過范德華力、氫鍵、色散力和其他化學物理作用使疏水性化合物或者表面活性劑吸附在固體表面上,也是導致固體表面帶電荷的另一重要原因。最典型的例子就是氧化硅表面上吸附腐殖酸而致使氧化硅表面帶有一定的電荷。圖5表示不同固體表面電荷對其所處體系的pH值改變的趨勢。結果表明,大多數固液界面都存在一定的表面電荷,且表面總電荷密度強烈依賴體系pH值。
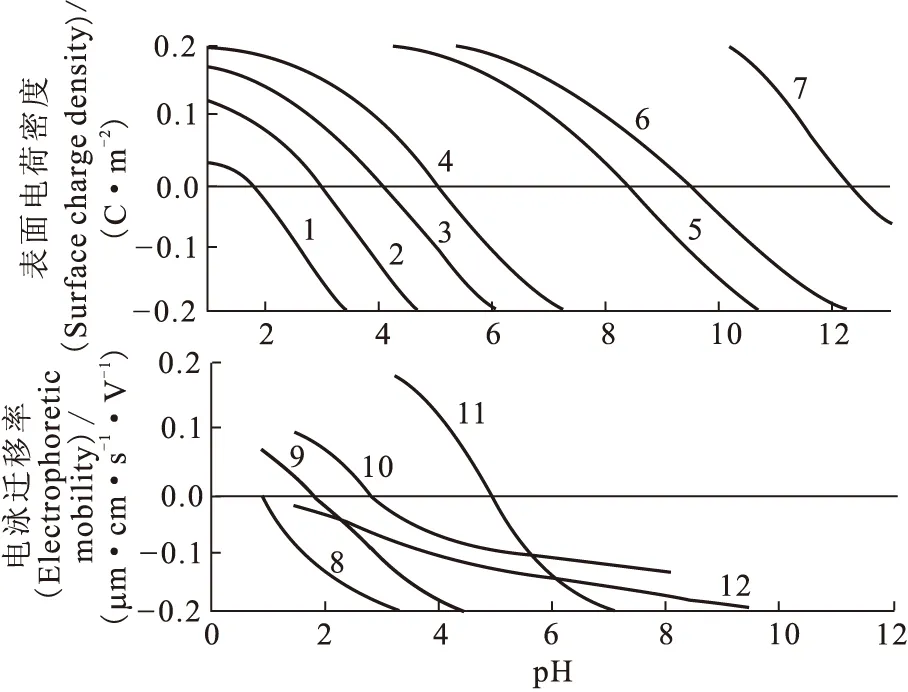
圖5 不同物質的表面電荷密度和電泳遷移率隨pH值改變趨勢[11]
1.2.2固體表面電荷密度和零電荷點 目前,連續電位滴定仍然是研究固體表面電位或電荷密度的重要方法之一。吸附劑表面位點密度、酸堿反應平衡常數以及零電荷點等參數都可以通過電位滴定獲得。而研究吸附劑表面酸堿性質是定量描述放射性核素在其表面上吸附和解吸機理的必要前提。Guo等[12]利用連續電位滴定的方法詳細探討了Na基膨潤土表面酸堿性質,發現同一離子強度下,樣品的回滴曲線和滴定曲線之間存在明顯的滴定滯后現象;而同一滴定方向(pH從4到9或pH從9到4)的滴定曲線間的差異則相對較小(圖6)。ICP-OES測量發現,滴定過程中溶液中Al(Ⅲ)的最大濃度為2.5×10-5mol/L,說明礦物溶解作用對滴定結果的影響很小,且礦物溶解似乎也不是滴定滯后的主要原因。滴定滯后的最主要原因尚不清楚,仍有待進一步研究。Duc等[13]研究了樣品的儲存條件、CO2、固液比、滴加時間間隔、離子強度等因素對黏土礦物尤其是蒙脫石滴定的影響,發現采用濕法保存樣品、惰性氣體保護(消除CO2的影響)、縮短滴加時間間隔(約為10 min)等滴定策略可在一定程度上消除滴定滯后作用,提高測量的重現性和準確度。Na基膨潤土表面過剩質子量(ΔQH)和Na基凹凸棒石表面電荷密度隨pH變化示于圖7。由圖7(a)可知,不同離子強度下的3條滴定曲線沒有交點且幾乎平行,零電荷點隨離子強度的減小而增大,這與文獻中Na基蒙脫石的連續滴定結果一致[13-14]。但Fan等[15]利用連續電位滴定計算Na型凹凸棒石黏土表面零電荷點時卻發現3種離子強度下滴定曲線幾乎交于一點(pH≈6.02)。目前,固體表面的零電位點的表征方法有很多,除連續電位滴定以外,還有固體添加法[16]、電泳遷移率法(electrophoretic mobility)[17]和質量滴定法等[18]。錢麗娟等[18]利用質量滴定的方法求得3種磷酸鹽的零電荷點(圖8)。隨著固液比逐漸增大,pH逐漸向一個定值接近。pH變化方向與起始的pH有關,最后趨向于一個交點,該點即為固體材料的pHpzc點。由質量滴定可知,ZrP2O7、Zr2O(PO4)2和Th4(PO4)4P2O7三種磷酸鹽的pHpzc分別為3.5、4.2和6.8。
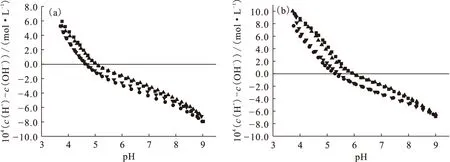
圖6 金川Na基膨潤土在0.1(a)、0.01(b)mol/L NaCl溶液中的酸堿滴定曲線
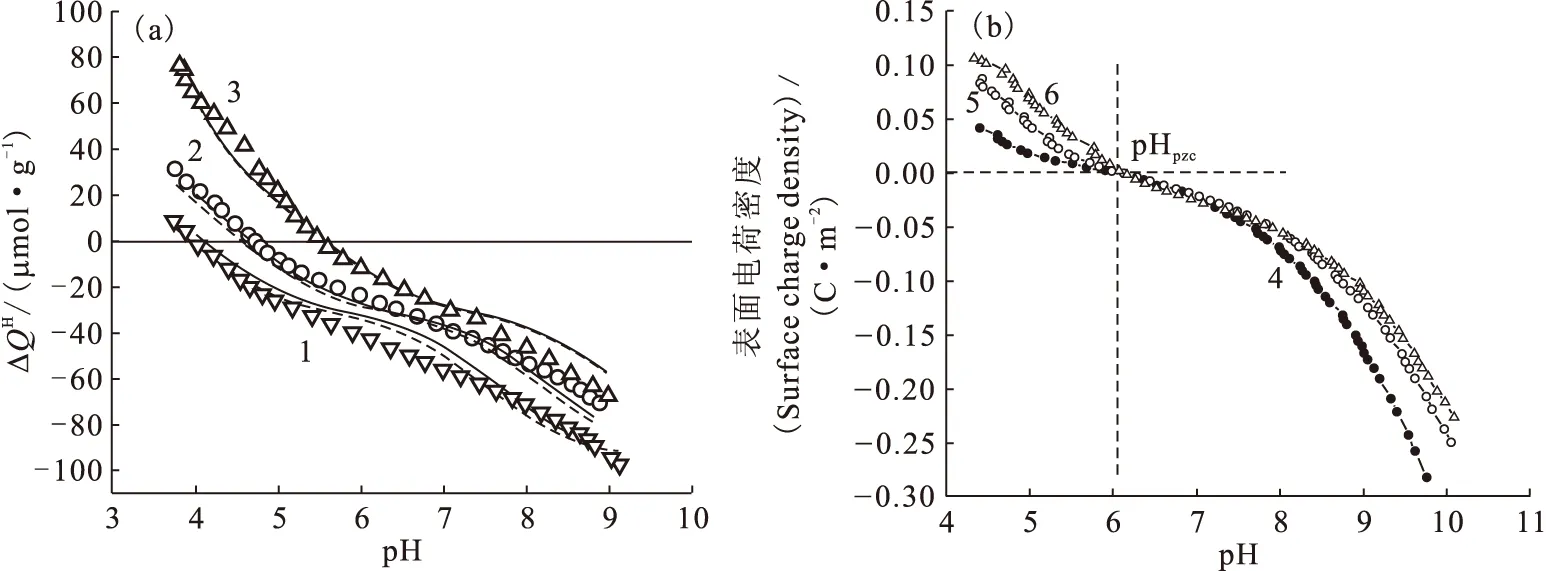
圖7 Na基膨潤土表面過剩質子量(a)[12]和Na基凹凸棒石表面電荷密度(b)[15]與pH關系圖
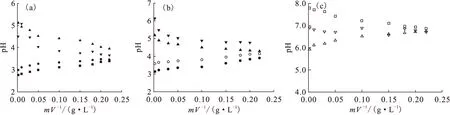
圖8 ZrP2O7(a)、Zr2O(PO4)2(b)、Th4(PO4)4P2O7 (c)的質量滴定曲線[18]
1.2.3表面配位模型 1998年Kraepiel等[19]提出兩種表面配位模型以解釋黏土礦物在不同離子強度下的平行滴定曲線。將黏土礦物看作一種不能穿透的固體物質,假設存在兩種表面吸附位點:?X-和?S—OH,其中?X-位點所帶的電荷與pH無關(永久性電荷),?S—OH位點所帶電荷來自于表面質子化和去質子化反應,礦物的表面電荷密度為永久性電荷和可變電荷之和。礦物表面電荷和表面電勢的關系可用Gouy-Chapman 理論計算[20]。假設:(1)蒙脫石表面除了層間吸附位點?X-(永久負電荷吸附位點),還存在兩類邊緣吸附位點,?Al—OH和?Si—OH,且N(?SiOH)/N(?AlOH)=2.0;(2)?Al—OH發生質子化和去質子化反應,而?Si—OH只發生去質子化反應[13];(3)Na基膨潤土層間位點?X-完全被Na+占據,?X-位點的容量由測得的陽離子交換容量(CEC)值來確定。金川Na基膨潤土表面發生的表面反應可用下列方程式表達:

(5)
(6)
(7)
(8)
(9)
依據上述表面反應,Na基膨潤土表面質子過剩ΔQH可以表示為:
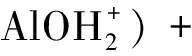
c(?XH)-c(?AlO-)-c(?SiO-))
(10)
其表面電荷ΔQcharge(mol/g)可以表示為:
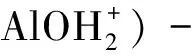
c(?AlO-)-c(?SiO-)-c(?X-))
(11)
其表面電荷密度σ(C/m2)為:
(12)
式中:F是法拉第常數,96 485 C/mol;s是Na基膨潤土的比表面積,m2/g;對于浸于對稱電解質溶液的膨潤土,其表面電勢(ψ,V)計算公式為:
σ=(8RTεε0ce×103)1/2sin(ZeψF/2RT)
(13)
式中:ε是水的介電常數(25 ℃為78.5 C/(V·m));ε0為真空介電常數,8.854×10-12C/(V·m);ce是背景電解質濃度;Ze是背景電解質的化合價。反應 (5)~(9)的本征平衡常數可表示為:
(14)
(15)
(16)
(17)
(18)
假設表面物種的活度系數為1,溶液中物種的活度系數用Davies 方程計算,擬合參數如下:?X-、?Al—OH(s)、?Al—OH(w)和?Si—OH的位點密度分別為1.16×10-5(623 meq/kg)、1.88×10-8、5.69×10-7、1.18×10-6mol/m2,比表面積為53.6 m2/g。其他列入表1。
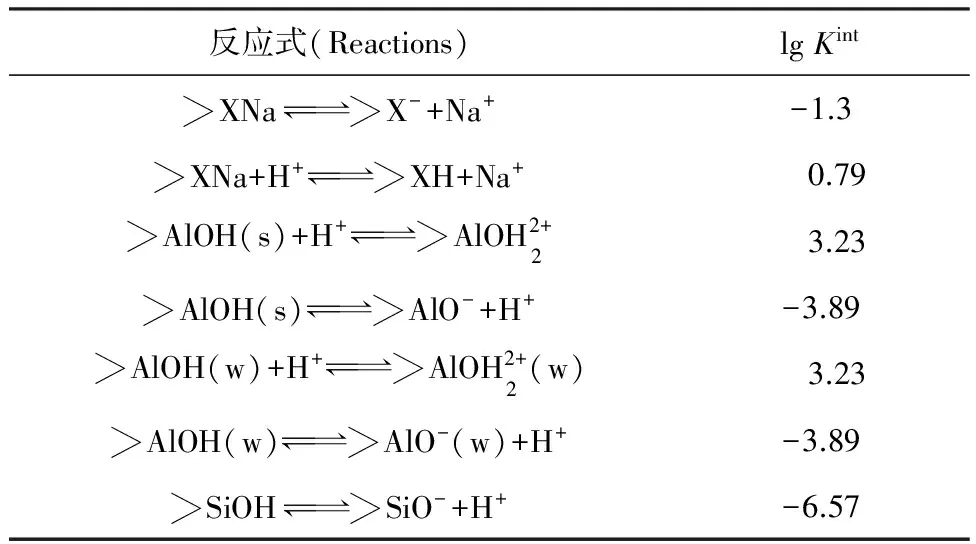
表1 FITEQL軟件擬合滴定曲線結果
2 影響放射性核素在固液界面吸附的因素
影響金屬離子或有機質在固液界面吸附行為的主要因素包括:(1)吸附劑固體本身物理和化學性質,如等電點、顆粒尺寸、固體的孔徑大小、比表面積、表面官能基團等;(2)環境體系pH、離子強度、溫度、壓力、有機質、空氣中各種氣體的含量(如CO2)等對吸附質在環境介質中的物種分布、價態等都有較強的影響,另外這些因素同樣對固體表面電荷和電位有著至關重要的影響;(3)吸附質的存在形態、價態、與溶液中各組分的相互作用等(包括與陰離子的配合作用和陽離子的競爭作用等)均對放射性核素在吸附劑表面上的吸附產生重要的影響。
2.1 pH和離子強度的影響
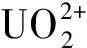
2.2 溫度影響
溫度是影響吸附質在固液界面上吸附行為的重要參數之一。通常溫度對放射性核素的吸附有正影響,即吸附體系溫度升高有利于放射性核素的吸附反應發生;但少數吸附質在固液界面上吸附與溫度卻成負相關,如Cs(Ⅰ)在天然高嶺土、斜發沸石和花崗巖上的吸附。溫度對吸附質在固液界面上的吸附影響可總結出以下經驗:(1)溫度升高使得吸附活化能增大,吸附熵增大,從而增大吸附質的最大吸附量;(2)吸附質在固液界面上的吸附過程可為放熱或吸熱過程,因此溫度升高時反應產物的量可能增加也可能減少;但值得注意的是物理吸附總是伴隨能量的釋放,即溫度升高總是削弱物理吸附反應;(3)當吸附質在固液界面上形成羥基配合物時,使得在不同pH下溫度對吸附的影響不同。
圖9 pH值對U(Ⅵ)在SiO2上的吸附影響(a)[21]和對Th(Ⅳ)在凹凸棒石上的吸附影響(b)[23]
2.3 腐殖質的影響
腐殖質是動植物經過長期的物理、化學、生物作用而形成的復雜天然有機物。天然水體系和土壤等都含有一定的腐殖質。腐殖質是大分子聚合物,化學結構復雜,其相對分子質量從幾百到幾千不等。Schnitzer和Schuppli[24-25]利用紅外光譜系統研究了土壤腐殖質的主要官能團組成,發現主要構成官能團有羧基、醇羥基、酚羥基、氨基、醌型羰基和酮型羰基等。不同提取劑提取的腐殖質在相對分子質量、芳化度和主要官能團方面均存在一些差異。各種官能團的含量在不同來源的腐殖酸中差異很大。就胡敏酸和富啡酸比較來看,富啡酸含羰基、醇羥基、酚羥基和酮羰基的量較胡敏酸多,而胡敏酸含醌羰基的量要比富啡酸高。一般情況下,在低pH值下,腐殖酸對放射性核素在固液界面上吸附起促進作用;而在高pH值條件下,則可能會抑制放射性核素的吸附。反之,由于腐殖酸沒有固定分子式和相對分子質量,其大分子結構形貌受到放射性核素、pH值、膠體顆粒等因素的影響,腐殖酸對放射性核素在氧化物-腐殖酸表面的作用機理影響不是非常清楚,還有待進一步研究。
3 微觀光譜技術在放射性核素吸附研究中的應用
3.1 XAFS技術應用
長期以來,由于實驗技術的局限性,放射性核素在固液界面體系中的化學行為研究多集中于宏觀熱力學和動力學層面上。20世紀80年代發展起來的同步輻射光源技術,使XAFS(X-ray absorption fine structure,X射線吸收精細結構譜)可以通過對固液界面體系中目標元素的原子周圍環境進行分子水平上的研究,而獲得無序表面組成和微觀結構信息,為從分子水平上研究放射性核素在固液界面上的吸附反應、揭示界面反應機理等提供了重要的技術支持。XAFS具有下列無可比擬的優勢:(1)EXAFS(擴展X射線吸收精細結構譜)現象來源于吸收原子周圍最近鄰的幾個配位殼層作用,決定于短程有序作用,且可用于非晶態物質的研究,得到吸收原子近鄰配位原子的種類、距離、配位數及無序度因子;(2)X射線吸收邊具有原子特征,可對不同元素的原子周圍環境分別進行研究;(3)利用強X射線或熒光探測技術可以測量到mg/kg濃度級別的環境樣品;(4)EXAFS可用于固體、液體、氣體等樣品,一般不需要高真空,且不損壞樣品。Fan等[15]利用EXAFS吸收譜證明了HA對Eu(Ⅲ)在凹凸棒石表面上吸附機理影響機制,發現不同HA的添加次序與Eu(Ⅲ)在固液界面上的微觀結構緊密相關。不同HA添加次序下,Eu(Ⅲ)在凹凸棒石黏土表面的徑向結構分布和k2χ(k)函數示于圖10。通過對第一配位殼層(Eu-O)進行擬合,得到相關的結構參數列于表2。由表2可知,對于batch 1 (ATP+HA-Eu(Ⅲ))樣品,原子間距d(Eu-O)≈0.241 1 nm(N=11.91,σ2=0.016 3);bacth 2(Eu(Ⅲ)+HA-ATP)樣品的原子間距d(Eu-O)≈0.239 9 nm(N=11.66,σ2= 0.016 3);而batch 3 (ATP+Eu(Ⅲ)-HA)樣品的d=0.232 1 nm(N=8.866,σ2=0.005 3)。值得注意的是batch 3樣品結構參數與二元體系(Eu(Ⅲ)+ATP)非常相似(d(Eu-O)=0.231 4 nm,N=8.24,σ2=0.001 6);而batch 1和2的結構參數相似。此結果很好的說明了有機質的存在、體系pH值等因素對放射性核素在固液界面上的吸附機理有至關重要的影響。
3.2 XPS技術應用
單純從靜態批式實驗數據和假設模型很難獲得較準確的放射性核素在固液界面上的吸附物種和機理。XPS技術對固體表面元素分析具有高靈敏性等特點,因此,XPS技術可以很好的應用到放射性核素在固液界面上的吸附機理研究中,將XPS光譜、靜態宏觀實驗和表面配位模型等相結合,必將促進人們對放射性核素在固液界面上微觀吸附機理的進一步認識和理解。圖11為pH分別為4.5和6.0時,吸附在凹凸棒石黏土和ATP/IOM復合材料上的U(Ⅵ)的U4f譜圖。由圖11可知:pH=4.5時,在U(Ⅵ)-凹凸棒石黏土二元體系中含378.57 eV和382.02 eV處的兩種物種;而在U(Ⅵ)-ATP/IOM復合材料體系中則只有382.56 eV處的一種物種形成;當pH≈6.0時,在U(Ⅵ)-ATP/IOM復合材料體系中也存在兩種物種(378.35 eV和382.05 eV處)。這一結果為后續的模型擬合奠定了基礎。
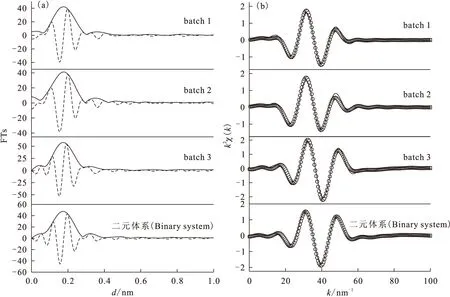
圖10 不同HA添加次序下Eu LⅢ吸收邊的徑向結構函數(a)和k2χ(k)函數(b)[15]
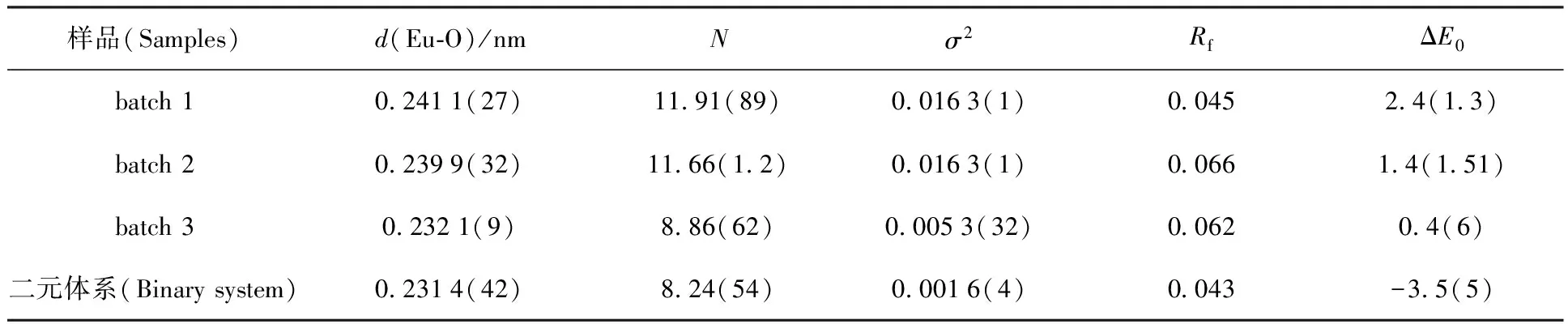
表2 不同HA添加次序的EXAFS擬合結果
注(Notes):t=(20±1)℃,c(NaClO4)=0.01 mol/L;d,原子間距離(Interatomic distance);N,配位數(Number of neighbor oxygen atoms);σ2,Debye-Waller因子(Factor);ΔE0,能量位移(Energy shift);Rf,殘留因子(Residual factor),Rf=∑k(k2xexp-k2xcalc)/∑k(k2xexp)
3.3 其他光譜技術應用
除了上述XAFS和XPS等光譜技術可以對放射性核素在固液界面上的微觀結構和吸附機理進行原子或分子水平上的分析外,X射線能量色散譜(EDS)、時間分辨激光熒光光譜(TRLFS)、激光誘導熒光法(LIF)、紅外(FTIR)、T-FTIR(透射)、漫反射(DR-FTIR)和衰減反射(ATR-FTIR)等光譜在固液界面上吸附的微觀機理和結構研究方面同樣有著重要的應用。SEM-EDS面分布是一種表面靈敏和直觀的分析方法,能夠直接得到目標元素在表面上的分布情況和在表面上的含量等信息。圖12為不同背景電解質條件下吸附樣品對Cs(I)吸附后的EDS分析。由圖12可知:Cs主要是均勻分布在土壤顆粒的基面(basal plane)和邊位點(frayed edge site,FES),說明Cs在土壤表面沒有特定吸附區域。這種現象表明Cs(Ⅰ)在石灰性土壤上的吸附主要依靠靜電作用的離子交換和范德華力。Cs的百分比隨背景電解質陽離子的改變而發生規律性變換,陽離子對Cs的競爭能力遵從Mg2+>Ca2+≈ Na+的順序。
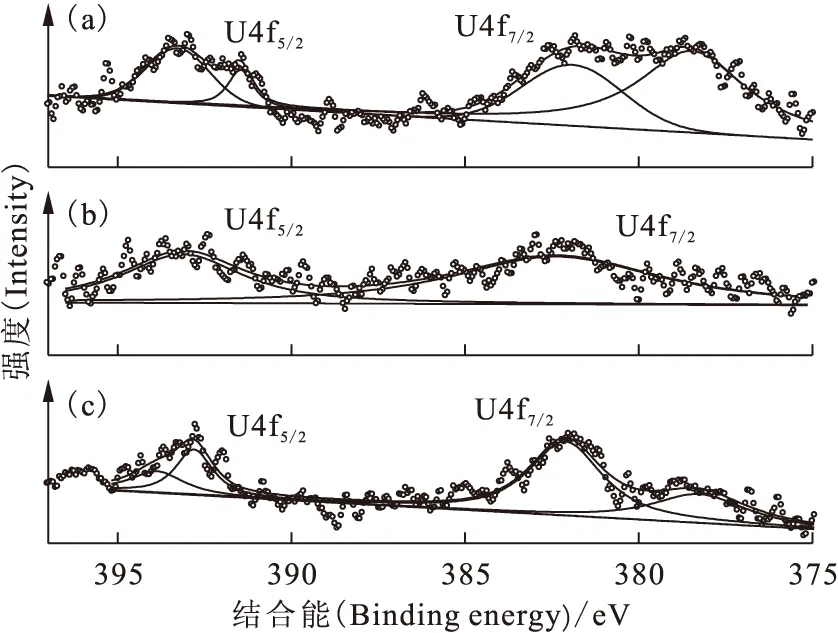
圖11 U(Ⅵ)在凹凸棒石黏土和ATP/IOM復合材料上的XPS U4f光譜[3]
圖13為Eu(Ⅲ)在三種溶液中的熒光強度衰減曲線。在氧化鋁存在下,半衰期110 μs 表明Eu(Ⅲ)的第一配位殼層含9個水分子,此結果與Eu3+離子在溶液中周圍有9個水分子完全吻合,結果表明,在氧化鋁溶液中Eu(Ⅲ)主要以水合Eu3+離子存在。210 μs表示Eu(Ⅲ)的第一配位殼層含4個水分子,說明氧化鋁存在下,除Eu(Ⅲ)以水合離子存在于溶液中之外,還有部分Eu(Ⅲ)在失去5個水分子后被吸附到氧化鋁顆粒表面上。在胡敏酸存在下,Eu(Ⅲ)只有一個熒光衰減時間,說明Eu(Ⅲ)與胡敏酸形成了一種穩定的配合物。在胡敏酸與氧化鋁共同存在下,Eu(Ⅲ)則有兩個不同的衰減時間,表明此時Eu(Ⅲ)形成了兩種不同形態的配合物,Eu(Ⅲ)的第一配位殼層含5個水分子,說明Eu(Ⅲ)吸附在氧化鋁的表層,在實驗條件(如pH值、離子強度等)改變時有可能從氧化鋁表層被解吸下來;而Eu(Ⅲ)第一配位殼層有2個水分子,說明Eu(Ⅲ)吸附在氧化鋁的內層,即使實驗條件改變也很難從氧化鋁上被解吸下來,此吸附屬于不可逆吸附。

圖12 不同條件下Cs在石灰性土壤表面分布
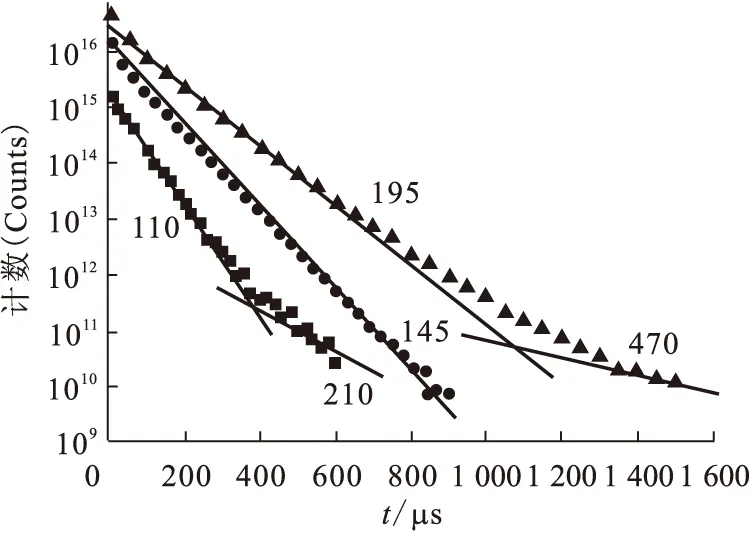
圖13 Eu(Ⅲ)在三種溶液中的熒光強度隨時間的變化關系[26]
4 放射性核素在固液界面的吸附機理
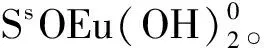
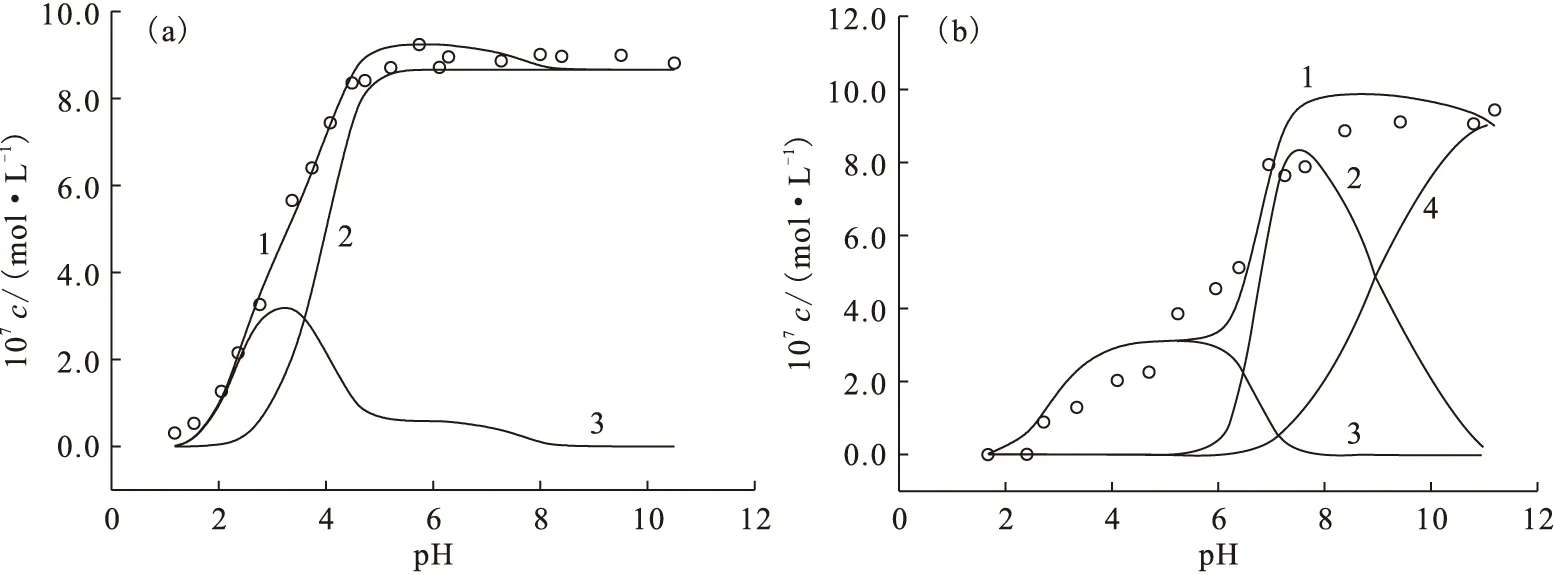
圖14 Eu(Ⅲ)在凹凸棒石表面上的吸附種態隨pH值的變化
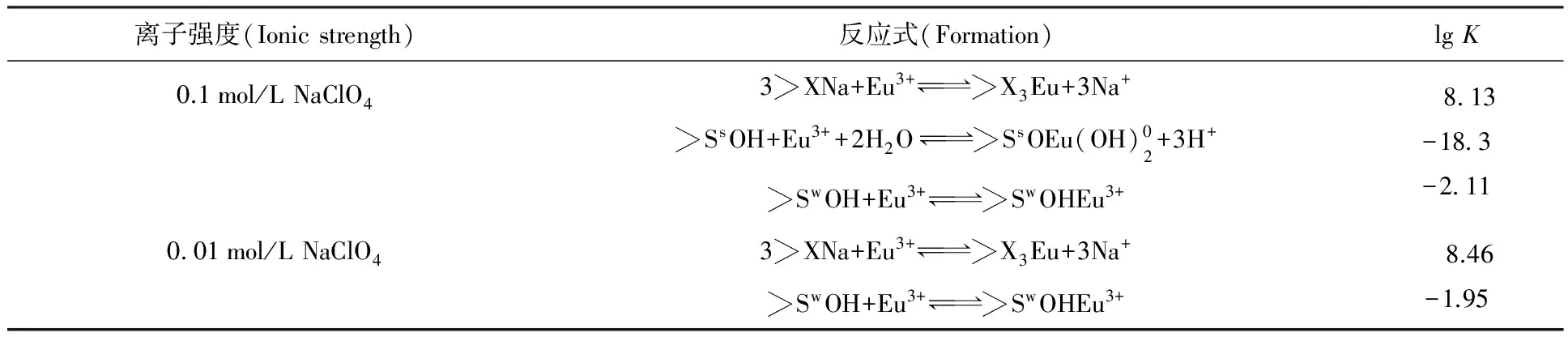
表3 Eu(Ⅲ)在凹凸棒石表面上的吸附模型
5 結 論
本文主要以放射性核素在氧化物、磷酸鹽和黏土表面上的吸附研究為例,綜述了放射性核素在固體吸附劑表面上的宏觀吸附實驗和微觀表征方法。影響放射性核素在固液界面上吸附的因素多且復雜,如pH值、離子強度、溫度和有機質等。因此選擇合適、正確的吸附模型對放射性核素在固液界面上的吸附研究具有重要的意義。表面配位模型在放射性核素吸附研究領域中取得了很大的成功,這主要是因為其模型能方便、合理地描述和解釋實驗數據。不同吸附材料由于其自身的結構、形貌和電荷等性質的差異,在微觀吸附種態和吸附機理的探討和論證中,還必須采用先進的光譜技術對微觀吸附過程進行分子水平上的研究,如EXAFS、XPS、TRLFS和EDS等對放射性核素在氧化物、磷酸鹽和黏土表面上吸附機理的探討。然而,這些分析技術在環境樣品的研究中仍然受到諸多因素的限制,仍需要探索和發展新的研究方法和手段,為得到正確和真實的吸附機理研究提供直接的、關鍵性的證據。
[1]Wang Y Q,Fan Q H,Li P,et al.The Sorption of Eu(Ⅲ)on Calareous Soil:Effects of pH,Ionic Strength,Temperature,Foreign Ions and Humic Acid[J].J Radioanal Nucl Ch,2011,287:231-237.
[2]Fan Q,Shao D,Lu Y,et al.Effect of pH,Ionic Strength,Temperature and Humic Substances on the Sorption of Ni(II)to Na-Attapulgite[J].Chem Eng J,2009,150(1):188-195.
[3]Fan Q H,Li P,Chen Y F,et al.Preparation of Attapulgite/Iron Oxide Magnetic Composites and Application in Removal U(Ⅵ)From Aqueous Solution[J].J Hazard Mat,2011,192(3):1 851-1 859.
[4]Zhao G X,Zhang H X,Fan Q H,et al.Sorption of Copper(Ⅱ)Onto Super-Adsorbent of Bentonite-Polyacrylamide Composites[J].J Hazard Mat,2010,173(1-3):661-668.
[5]Chen C L,Wang X K,Nagatsu M.Europium Adsorption on Multiwall Carbon Nanotube/Iron Oxide Magnetic Composite in the Presence of Polyacrylic Acid[J].Environ Sci Technol,2009,43:2 362-2 367.
[6]Oonk S,Slomp C P,Huisman D J,et al.Geochemical and Mineralogical Investigation of Domestic Archaeological Soil Features at the Tiel-Passewaaij Site,the Netherlands[J].J Geochem Explor,2009,101:155-165.
[7]Fan Q,Li Z,Zhao H,et al.Adsorption of Pb(Ⅱ)on Palygorskite From Aqueous Solution:Effects of pH,Ionic Strength and Temperature[J].Appl Clay Sci,2009,45(3):111-116.
[8]Zhang Y Y,Zhao H G,Fan Q H,et al.Sorption of U(Ⅵ)Onto a Decarbonated Calcareous Soil[J].J Radioanal Nucl Ch,2011,288(2):395-404.
[9]Anderson M A,Rubin A J.Adsorption of Inorganic at Solid-Liquid Interface[M].Ann Arbor,Michigan:Ann Arsor Sci,1981,Charp.1.
[10]陶祖貽,杜金洲.氧化物/水界面上的表面絡合模型[J].離子交換與吸附,1994,10(2):112-118.
[11]Stumm W.Chemistry of the Solid-Water Interface:Processes at the Mineral-Water Interface in Natural Systems[M].New York:Wiley-Interscience,1992:43.
[12]Guo Z J,Xu J,Shi K L,et al.Eu(Ⅲ)Adsorption/Desorption on Na-Bentonite:Experimental and Modeling Studies[J].Colloid Surf A,2009,339(1-3):126-133.
[13]Duc M,Gaboriaud F,Thomas F.Sensitivity of the Acid-Base Properties of Clays to the Methods of Preparation and Measurement:2.Evidence From Continuous Potentiometric Titrations[J].J Colloid Interf Sci,2005,289(1):148-156.
[14]Tertre E,Berger G,Simoni E,et al.Europium Retention Onto Clay Minerals From 25 to 150 ℃:Experimental Measurements,Spectroscopic Features and Sorption Modelling[J].Geochim Cosmochim Acta,2006,70(18):4 563-4 578.
[15]Fan Q H,Tan X L,Li J X,et al.Sorption of Eu(Ⅲ)on Attapulgite Studied by Batch,XPS,and EXAFS Techniques[J].Environ Sci Technol,2009,43(15):5 776-5 782.
[16]Srivastava V C,Mall I D,Mishra I M.Adsorption of Toxic Metal Ions Onto Activated Carbon Study of Sorption Behaviour Through Characterization and Kinetics[J].Chem Engin Process,Process Intensification,2008,47:1 269-1 280.
[17]Drot R,Lindecker C,Fourest B,et al.Surface Characterization of Zirconium and Thorium Phosphate Compounds[J].New J Chem,1998,22:1 105-1 109.
[18]錢麗娟,胡佩卓,蔣正江,等.pH、富里酸和溫度對鈾酰在ZrP2O7上的吸附影響[J].中國科學 化學,2010,40:1 712-1 720.
[19]Kraepiel A M L.On the Acid-Base Chemistry of Permanently Charged Minerals[J].Environ Sci Technol,1998,32:2 829-2 838.
[20]Hoch M,Weerasooriya R.New Model Calculations of pH-Depending Tributyltin Adsorption Onto Montmorillonite Surface and Montmorillonite-Rich Sediment[J].Environ Sci Technol,2004,39(3):844-849.
[21]Zhang H,Wen C,Tao Z,et al.Effects of Nitrate,Fulvate,Phosphate,Phthalate,Salicylate and Catechol on the Sorption of Uranyl Onto SiO2:A Comparative Study[J].J Radioanal Nucl Ch,2011,287:13-20.
[22]許君政,范橋輝,白洪彬,等.離子強度、溫度、pH和腐殖酸濃度對Th(Ⅳ)在凹凸棒石上吸附的影響[J].核化學與放射化學,2009,31(3):179-185.
[23]Wu W S,Fan Q H,Xu J Z,et al.Sorption-Desorption of Th(Ⅳ)on Attapulgite:Effects of pH,Ionic Strength and Temperature[J].Appl Radiat Isot,2007,65(10):1 108-1 114.
[24]Schnitzer M,Khan S U.Humic Substances:Chemistry and Reactions In:Soil Organic Matter[M].Amsterdam:Elsevier,1978:1-64.
[25]Schnitzer M,Schuppli P.Method for the Sequential Extraction of Organic Matter From Soils and Soil Fractions[J].Soil Sci Soc Amer J,1989,53(5):1 418-1 424.
[26]王祥科,鄭善良.熒光衰減光譜法研究Eu(Ⅲ)在氧化鋁表面的化學形態[J].核化學與放射化學,2005,27(2):108-112.
[27]Naveau A,Monteil-Rivera F,Dumonceau J,et al.Sorption of Europium on a Goethite Surface:Influence of Background Electrolyte[J].J Contam Hydrol,2005,77(1-2):1-16.
[28]Bradbury M H,Baeyens B.Sorption of Eu on Na- and Ca-Montmorillonites:Experimental Investigations and Modelling With Cation Exchange and Surface Complexation[J].Geochim Cosmochim Acta,2002,66(13):2 325-2 334.
猜你喜歡
艦船科學技術(2022年16期)2022-09-22 02:15:00
北京航空航天大學學報(2021年6期)2021-07-20 07:23:54
當代陜西(2020年13期)2020-08-24 08:22:02
制造技術與機床(2017年5期)2018-01-19 02:49:17
制造技術與機床(2017年11期)2017-12-18 06:47:29
金秋(2017年4期)2017-06-07 08:22:16
蘇州科技大學學報(自然科學版)(2017年1期)2017-03-20 15:25:18
中國材料進展(2016年10期)2016-12-26 06:50:20
濰坊學院學報(2016年2期)2016-12-01 13:00:11
新聞傳播(2015年11期)2015-07-18 11:15:04
