基于EST序列的煙草cSNP發掘
2012-01-17 05:36:52龔達平李鳳霞王衛鋒劉貫山孫玉合
中國煙草科學 2012年6期
關鍵詞:煙草
龔達平,王 魯,李鳳霞,王衛鋒,劉貫山,孫玉合
(農業部煙草類作物質量控制重點開放實驗室,中國農業科學院煙草研究所,青島 266101)
單核苷酸多態性(single nucleotide polymerphisms,SNP)主要是指在基因組水平上由單個核苷酸的變異所引起的DNA序列多態性。SNP在基因組中遺傳穩定、分布廣泛、多態性高、易實現自動化檢測,被認為是最具應用潛力的第三代分子標記。除在遺傳分析以及作物分子標記輔助育種中有著廣闊的應用前景外,其在構建高密度遺傳圖譜,整合物理圖譜,復雜遺傳性狀以及基因組進化研究等方面有著重要的應用價值[1-3]。
近幾年來,新型大規模、高通量SNP發掘方法不斷涌現。如DNA列陣微測序法、動態等位基因特異雜交法、寡聚核苷酸特異連接法、DNA芯片以及基質輔助激光解析電離飛行時間法等。特別是以邊合成(或連接)邊測序為特征的新一代測序技術的出現,如 Roche (454)GSFLX sequencer、Illumina genome analyzer (Solexa)、Applied Biosystems SOLiD sequencer、HeliScope Sequencer等[4],使基因組學和功能基因組學進入了一個低成本、大規模、高通量測序的時代。但是,普通煙草(N.tabacum)為異源四倍體(TTSS),基因組龐大,重復序列高,且T、S染色體組間存在很高的同源性。因此對煙草全基因組進行測序和組裝是一項非常艱巨的工作。美國北卡州立大學僅對普通煙草基因組的基因富集區進行了測序。傳統分子標記的開發耗時費力,而目前在公共數據庫 GenBank中存儲了超過30萬條來源于不同品種的普通煙草EST序列,這為我們進行基于 EST數據的高度冗余性來發掘SNP提供了豐富的數據資源。特別是編碼區 SNP(coding SNP,cSNP)位于外顯子內,有可能與基因功能直接相關,因此它們在煙草的重要性狀研究中具有重大的應用價值[5-6]。
目前煙草基因組及SNP發掘才剛剛開始。煙草SNP研究大多集中于對單個基因的SNP分析上,只有歐洲煙草EST測序項目在開展普通煙草EST測序工作中發掘了 74 SNPs和 38 InDels(www.ESTobacco.info)。本研究中,我們利用公共數據庫中的EST序列,在無需序列質量文件的條件下保守的挖掘cSNP,并對這些cSNP位點的可信度進行評估。
1 材料與方法

2 結 果
2.1 序列組裝
從GenBank中共檢索到煙草EST 317 175條。這些序列所產生的cDNA文庫來源的品種和組織器官以及發育時期如表1。共組裝成35 504個contig和109 550個singleton(表2)。包含至少4個讀長的contig共計15 429個。

表1 煙草EST數據來源的材料和組織Table 1 Cultivar and tissue of tobacco EST data
2.2 鑒定候選SNP
在包含至少4個以上reads的15 429個重疊群中,有7088個重疊群包含了冗余度為2以上的候選SNP共53 477個。不同大小的重疊群中包含候選 SNP的重疊群占該大小重疊群總數的比例如圖1。圖中只列出了重疊群大小為25個reads以下的分布。整體上隨著contig所包含reads數量的增加,包含SNP的contig占該大小重疊群總數的比例也迅速增加。如4個reads的重疊群中包含SNP的重疊群占 13.2%,當讀長數目增加到 13時,比例達到80%,而24個reads以上的重疊群中則達到94%以上,44個以上reads的重疊群全部包含SNP。
如圖2,所檢測到的42 420個候選SNP,其豐度也是隨重疊群的增大而增加。SNP的分值也隨著比對序列數目的增加而增加。平均300 bp的序列長度出現一個SNP位點。

表2 煙草EST數據不同大小重疊群的數量分布Table 2 Contigs profile of tobacco EST data

圖1 不同大小的重疊群中包含候選SNP的重疊群比例Fig.1 Abundance of tobacco EST contigs containing candidate SNPs in relation to contig size.
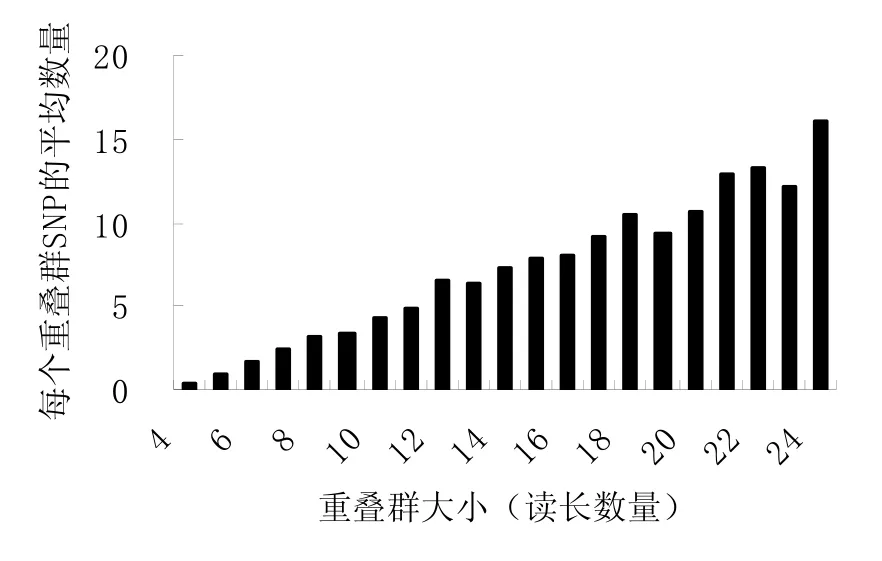
圖2 不同大小重疊群包含SNP的豐度Fig.2 Abundance of candidate SNPs identified within contigs in relation to contig size for tobacco EST data
2.3 堿基變化分析
由表3可見,候選的SNP歸類為轉換(C/T或G/A)或顛換(C/G,A/T,C/A 或 T/G)。轉換為26 582個,顛換為15 838個,轉換所占的比例比顛換要大。SNP的高頻轉換在其他研究中也有發現,反映了甲基化后C/T的高頻突變。
2.4 插入/刪除分析
試驗中鑒定了冗余值大于2的插入/刪除(Indels)10 057個。不同長度的Indels的分步頻率如表4。表4可見Indel頻率隨著長度的增加而降低。不管是單個還是多個堿基的 Indel,都表現出 A/T偏好(表5)。
2.5 單體型分析
除了SNP冗余度分值外,另一個SNP可信度的評價標準是通過基于對 SNP位點模式的共分離計算得到的。序列間的SNP代表了兩個基因(旁系同源或直系同源)間的分化,被期望是共分離的,而在多位點上定義一個單倍型可以在一定程度上避免測序錯誤的隨機發生。如表6和表7所示,在該重疊群中共鑒定7個候選SNP構成單體型,6個SNP位點冗余度為5,并具有較高的共分離分值和加權共分離分值,說明這些SNP位點具有較高的可信度。
比較序列來源的品種信息可以鑒定預測直系同源基因(表6)。通過隨機檢測100個單體型可以觀察到近一半的單體型在同品系中的分離表明在單個品系中多基因的表達。尤其是在重疊群的數量和SNP的數量均比較多的情況下,這些SNP最有可能是來源于普通煙草T和S兩個基因組中相似度極高的同源基因。

表3 煙草候選SNP的替換頻率Table 3 Nucleotide substitution frequencies for candidate SNPs identified in tobacco EST data

表4 候選Indel數量分布Table 4 Prevalence of candidate indels identified in tobacco EST data

表5 單個和2個堿基插入刪除頻率Table 5 The frequency of single and dinucleotide indel sequences predicted from the alignment of tobacco EST sequences
表6 重疊群包含的reads的文庫和品種信息Table 6 The contig contained the sequences with ID, name and line derived from the Genbank annotation.
3 討 論
SNP是一種可用作分子遺傳分析的新型分子標記。同其他分子標記一樣,其開發雖然可以自動化、規模化,但也很昂貴費力。而通過挖掘序列數據資源得到豐富的SNP是最為廉價的方法。通過篩選高質量的測序數據來發現具有生物學相關的SNP,不可避免地受到測序錯誤的干擾。以前主要依賴測序數據的質量文件來過濾 SNP,低質量的SNP位點可能是測序錯誤,而非真正的SNP位點。公共數據庫中的高冗余數據往往缺乏質量文件。EST數據的冗余性允許對SNP位點的發生頻率,即冗余分值,進行評估。通過檢測SNP的分值大于2或更大,可以排除大多數的測序錯誤。雖然遺漏了一些只出現過一次的真實SNP,但數據的高冗余度允許我們從各種不同來源的數據中快速鑒定大量的SNP[9-10]。
應用這種方法我們挖掘了煙草EST數據中的SNP。目前煙草的EST數據超過30萬條,通過嚴格的參數限制移除了54%包含少于4個讀長的重疊群。大量包含4個以上讀長重疊群保證了冗余度,增加了 SNP的可信度。從結果中可以看出,包含SNP的重疊群的比例和每個重疊群中 SNP的數目都隨著數據量增大而增加。這里我們鑒定了42 420個候選的普通煙草SNP,這仍只是煙草遺傳變異的一小部分。
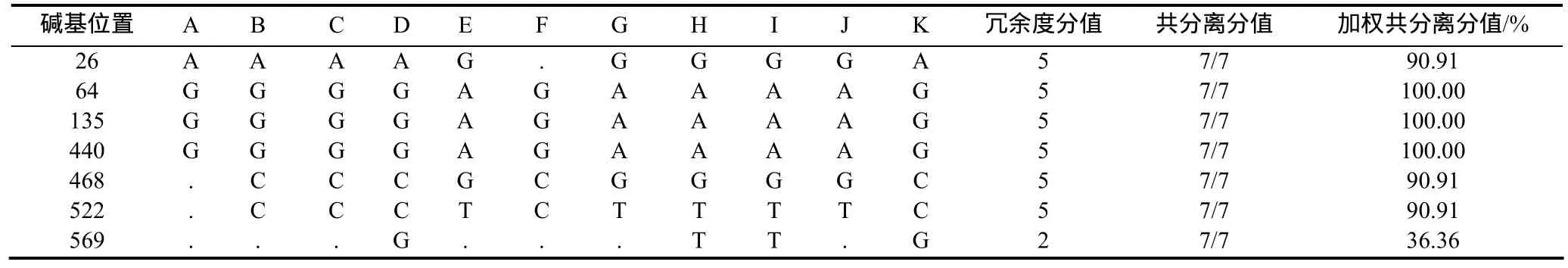
表7 重疊群中SNP統計Table 7 SNP summary in the contig
采用冗余度排除測序錯誤是有效的,但非隨機的測序錯誤也會造成兩次相同的序列錯誤。為了區分這些序列錯誤,需要進一步的SNP可信度衡量標準。代表兩個同源基因分化的SNP會共分離形成單體型。基于一個比對中多個SNP位點的SNP模式的頻率的共分離分值,允許鑒定非共分離的SNP。通過對SNP位點的數量和缺失數據的加權,能比較比對間的共分離分值。SNP分值和共分離分值一起提供評價SNP真實性的準確度,那些高冗余分值和共分離分值的SNP可能代表了真實的SNP位點。
SNP可以區分同一基因組中的復制基因(旁系同源基因)或不同品種中的直系同源基因。來自同一個品系的序列中的SNP,他們一定是由于基因復制和旁系同源基因的表達。通過隨機檢查100個單體型的分析表明,來自同一品種的多個基因間的SNP比例達到一半,這反映了普通煙草異源四倍體的起源,并且兩個基因組高度同源。估計兩個基因組間的相似程度是避免全基因組測序組裝錯誤所必要的。因此,大量的SNP來區分煙草中的旁系同源基因有助于基因組序列的組裝。
4 結 論
基于煙草EST數據的高度冗余性,317 175條普通煙草EST序列拼接成35 504個contig和109 550個singleton。其中,包含至少4個讀長的contig共計15 429個,說明公共數據庫中煙草EST數據足夠開發大量的SNP標記。共鑒定出53 477個冗余度大于2的候選SNP,煙草中平均300 bp出現一個SNP位點。其中,SNP的轉換率大于顛換,插入/刪除表現出A/T偏好。結合SNP位點的冗余度和共分離分值來評價其可信度,保證了所獲得的SNP位點具有較高的可信度。這些高質量SNP標記的發掘為進一步的煙草功能基因研究和分子育種奠定了基礎。
[1]Gupta P K, Roy J K, Prasad M.Single nucleotide polymorphisms: a new paradigm for molecular marker technology and DNA polymorphism detection with emphasis on their use in plants[J].Curr Sci., 2001, 80:524-535.
[2]Rafalski A. Applications of single nucleotide polymorphisms in crop genetics[J].Curr Opin Plant Biol.,2002, 5(2): 94-100.
[3]Syvanen A C. Genotyping single nucleotide polymorphisms[J].Nat Rev Genet., 2001, 2(12):930-942.
[4]Voelkerding K V, Dames S A, Durtschi J D.Next Generation Sequencing: From Basic Research to Diagnostics[J].Clin Chem., 2009, 55(4):641-658..
[5]Picoult N L, Ideker T E, Pohl M G, et al.Mining SNPs from EST databases[J].Genome Res, 1999, 9(2):167-174.
[6]Lein W, Usadel B, Stitt M, et al.Large scale phenotyping of transgenic tobacco plants (Nicotiana tabacum) to identify essential leaf functions[J].Plant Biotechnol J.,2008, 6(3): 246-263.
[7]Huang X, Madan A.CAP3: A DNA Sequence Assembly Program[J].Genome Research, 1999, 9(9): 868-877.
[8]Barker G, Batley J, O’ Sullivan, et al.Redundancy based detection of sequence polymorphisms in expressed sequence tag data using autoSNP[J].Bioinformatics,2003, 19 (3): 421-422.
[9]Batley J, Barker G, O'Sullivan H, et al.Mining for Single Nucleotide Polymorphisms and Insertions/Deletions in Maize Expressed Sequence Tag Data[J].Plant Physiology, 2003, 132(1): 84-91.
[10]毛新國,湯繼鳳,周榮華,等.基于全長 cDNA序列的小麥 cSNP發掘[J].作物學報,2006,32(12):1836-1840.
猜你喜歡
奧秘(創新大賽)(2023年3期)2023-05-06 01:48:20
中國煙草學報(2019年5期)2019-11-14 07:54:12
首都公共衛生(2019年5期)2019-05-21 01:08:34
浙江中西醫結合雜志(2017年2期)2017-01-12 18:23:59
新聞傳播(2016年3期)2016-07-12 12:55:34
當代化工研究(2016年9期)2016-03-20 16:22:08
自動化博覽(2014年6期)2014-02-28 22:32:15
聲屏世界(2014年6期)2014-02-28 15:18:09
西南學林(2013年2期)2013-11-12 12:58:54
中國煙草學報(2012年5期)2012-04-12 06:21:18
