HPLC法測定咖啡酸及咖啡酸片的含量
2012-05-01 13:22:54李明霞張英芳黃書芹
藥學(xué)研究 2012年6期
李明霞,張英芳,黃書芹
(1.德州德藥制藥有限公司,山東 德州 253015;2.德州市立醫(yī)院,山東 德州 253012)
咖啡酸為有機酸及酚類原料,其制劑咖啡酸片為止血升白細胞藥,具有收縮增固微血管、提高凝血因子的功能、升高白細胞和血小板的作用。咖啡酸與咖啡酸片的質(zhì)量標(biāo)準(zhǔn)分別收載于國家食品藥品監(jiān)督管理局局頒藥品標(biāo)準(zhǔn)與衛(wèi)生部頒藥品標(biāo)準(zhǔn)(化學(xué)藥品與制劑)第一冊,均采用紫外-分光光度法(吸收系數(shù)法)測定其含量,由于紫外-分光光度法的影響因素較多,且供試品溶液制備過程較為繁瑣,本文參照咖啡酸局頒藥品標(biāo)準(zhǔn)中有關(guān)物質(zhì)測定方法,采用高效液相外標(biāo)法測定其含量,方法簡便,準(zhǔn)確。
1 儀器與試藥
1.1 儀器 島津 LC-10ATvp高效液相色譜儀,SPD-10Avp紫外檢測器,Anastar色譜工作站。
1.2 試藥 咖啡酸 (由德藥制藥有限公司提供,批號:111101、111201、111202);咖啡酸片(由德藥制藥有限公司提供,批號:111101、111102、111103);咖啡酸對照品(中國藥品生物制品檢定所,批號為110885-200102);試驗用水為注射用水,甲醇為色譜純,冰醋酸為分析純。
2 試驗方法與結(jié)果
2.1 色譜分析條件及系統(tǒng)適用性 色譜柱:Sepax Sapphire C18柱(4.6mm ×250mm,5μm);流動相:甲醇 -0.32% 醋酸溶液(45∶55);檢測波長:323 nm;流速:1.0mL·min-1;進樣量:10μL。理論板數(shù)按咖啡酸峰計算應(yīng)不低于3000。
2.2 溶液的制備
2.2.1 對照品溶液的制備 取咖啡酸對照品12.5mg,精密稱定,置50mL量瓶中,加流動相溶解并稀釋至刻度,精密量取5mL,置25mL量瓶中,加流動相稀釋至刻度。
2.2.2 供試品溶液Ⅰ(咖啡酸)的制備 取咖啡酸12.5mg,精密稱定,置50mL量瓶中,加流動相溶解并稀釋至刻度,精密量取5mL,置25mL量瓶中,加流動相稀釋至刻度。
供試品溶液Ⅱ(咖啡酸片)的制備:取咖啡酸片20片,精密稱定,研細,取細粉適量(約相當(dāng)于咖啡酸12.5mg),精密稱定,置50mL量瓶中,加流動相溶解并稀釋至刻度,濾過,精密量取續(xù)濾液5mL,置25mL量瓶中,加流動相稀釋至刻度。
2.2.3 陰性對照溶液的制備 按咖啡酸片處方比例稱取輔料適量,同法制備空白溶液。
2.3 干擾試驗 在上述色譜條件下,取對照品、供試品和陰性對照溶液各10μL,注入液相色譜儀,記錄色譜圖。結(jié)果在對照品色譜峰保留時間相應(yīng)的位置上,供試品溶液有響應(yīng)的峰,陰性對照溶液無對應(yīng)峰,對樣品的測定無干擾,分離良好,見圖 1、2。
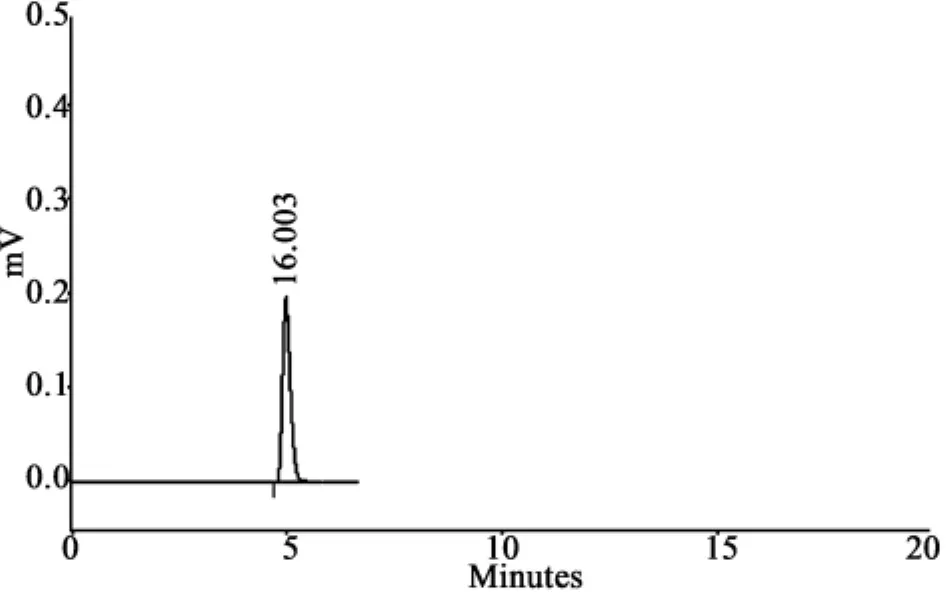
圖1 咖啡酸對照品色譜圖
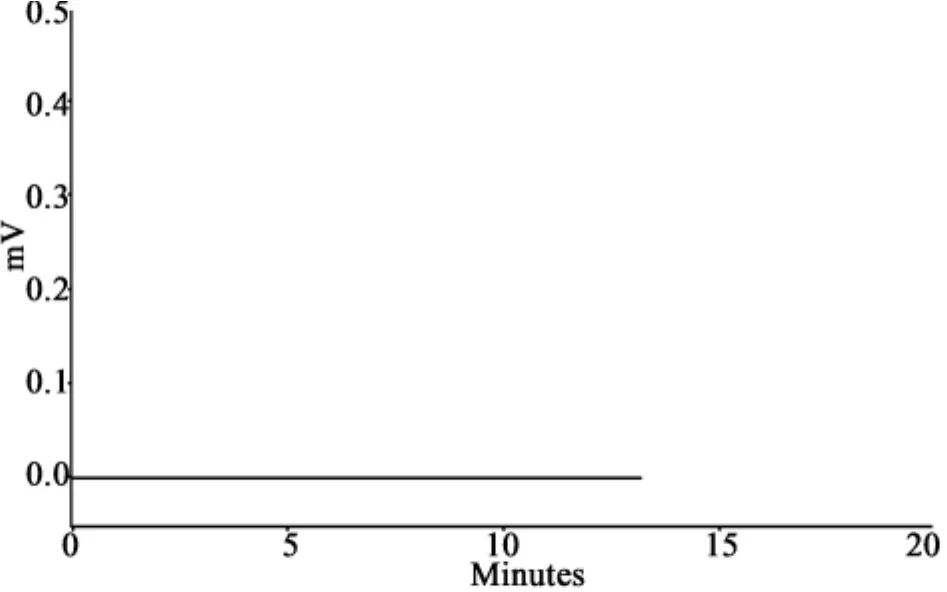
圖2 陰性對照溶液色譜圖
2.4 線性關(guān)系及范圍 取咖啡酸對照品12.5mg,精密稱定,置50mL量瓶中,用流動相溶解并稀釋至刻度,作為對照品儲備液。精密量取對照品儲備液 1.0、3.0、5.0、7.0、9.0mL,分別置25mL量瓶中,用流動相稀釋至刻度,搖勻,照“2.1”項色譜條件進行測定,記錄色譜圖,以對照品溶液濃度(C)為橫坐標(biāo),峰面積(A)為縱坐標(biāo),作線性回歸,得回歸方程A=47806C-5860.5,r=0.9998(n=5)。結(jié)果表明咖啡酸在 10 ~90 μg·mL-1范圍內(nèi),與峰面積呈良好的線性關(guān)系。
2.5 回收率試驗 分別取處方比例的咖啡酸對照品及空白輔料適量,照測定濃度的80%,100%,120%制備供試品溶液,每個濃度制備3份,照正文含量測定方法測定,將測得峰面積代入線性方程,計算供試品中咖啡酸的含量,回收率試驗結(jié)果見表1。
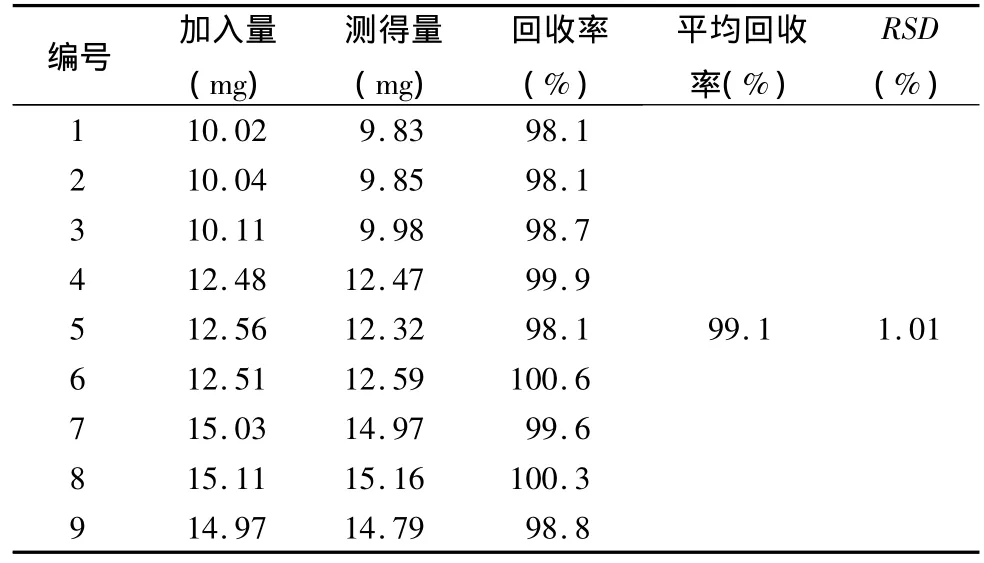
表1 回收率試驗結(jié)果
2.6 精密度試驗 取線性試驗中濃度為50 μg·mL-1的咖啡酸對照品溶液,重復(fù)進樣6次,按咖啡酸峰面積計算得RSD=0.67%(n=6),結(jié)果表明進樣精密度良好。
2.7 重復(fù)性試驗 取同一批號咖啡酸及咖啡酸片各6份,按正文方法測定,結(jié)果表明含量平均值分別為100.1%、99.6%,RSD 分別為0.86%、0.99%,表明本分析方法重復(fù)性較好。
2.8 穩(wěn)定性試驗 取同一供試品溶液,分別在0、2、4、6、8 h取10μL注入液相色譜儀,測定其峰面積,RSD=1.06%,表明本品溶液在8 h內(nèi)穩(wěn)定。
2.9 樣品測定 按正文擬定方法分別測定3批咖啡酸及咖啡酸片的含量,測定結(jié)果見表2。
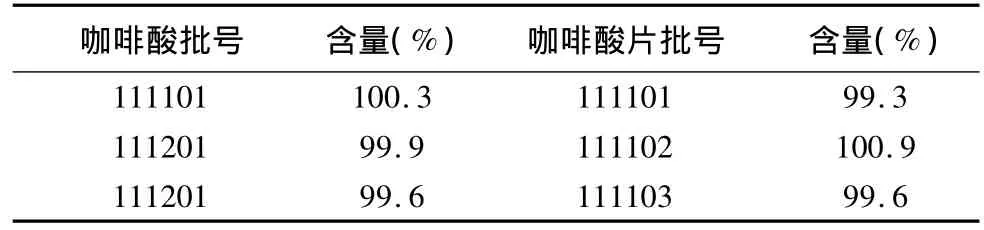
表2 咖啡酸及咖啡酸片樣品測定結(jié)果
3 討論
3.1 流動相、色譜柱的選擇均參照咖啡酸局頒質(zhì)量標(biāo)準(zhǔn)中有關(guān)物質(zhì)的測定。
3.2 檢測波長的選擇 取對照品溶液進行紫外掃描,結(jié)果咖啡酸在323 nm波長處有最大吸收,故選擇323 nm作為檢測波長,可用于含量測定。
3.3 本方法線性關(guān)系試驗、回收率試驗、精密度等試驗均良好,適用于咖啡酸及咖啡酸片的含量測定,方法簡單、精密度高、結(jié)果準(zhǔn)確可靠。
