化瘀無糖顆粒制備工藝的篩選
2012-08-05 01:27:26劉向紅陳金娜
山東醫藥 2012年46期
關鍵詞:工藝
劉向紅,陳金娜
(1山東大學齊魯醫院,濟南250012;2山東中醫藥大學)
化瘀顆粒為山東大學齊魯醫院自制制劑,由人參、蒲公英、漏蘆、水蛭等十三味中藥組成,用于腫瘤術后及放化療后,具有化瘀解毒、鎮痛及增強機體免疫力等作用。為了擴大適應證,使其可用于糖尿病患者和糖耐量異常人群,2011年8月~2012年5月,我們在原制劑基礎上進行一系列工藝改進,研制出了化瘀無糖顆粒。現將其制備工藝研究報告如下。
1 主要儀器及藥品
高效液相色譜儀(6CE,美國Waters公司)、Waters 2996檢測器、Empower600色譜工作站、分析天平(CP2250,奧豪斯國際貿易有限公司)、876-Ⅰ型真空干燥箱(南通科學儀器廠),旋轉蒸發器(RE-52A,上海亞榮生化儀器廠),貝利微粉機(BFMYT6B,濟南貝利粉技術工程有限公司)。咖啡酸對照品(批號110885-200102,購自中國食品藥品檢定研究院),糊精(東阿阿膠集團,批號20110115),阿斯巴甜(鄭州泰維生物技術有限公司)。均為藥用。化瘀顆粒所含的十三味中藥材均由濟南藥業集團購得,經山東中醫藥大學附屬醫院張學順主任藥師鑒定,均符合藥典規定。
2 方法與結果
2.1 化瘀無糖顆粒提取工藝的篩選 人參、水蛭制成超微粉,余蒲公英等11味中藥按傳統方法加水煎煮[1]。
2.1.1 試驗設計 按處方比例稱取上述藥材9份,每份試驗藥材重264 g。采用L9(34)正交試驗法[2](見表1),對提取效果影響較顯著的因素[加水量(A)、煎煮次數(B)、煎煮時間(C)]進行觀察,以咖啡酸含量和干膏得量(權重系數均為0.5,進行加權求和計算綜合評分,滿分10分)作為指標進行綜合評價。
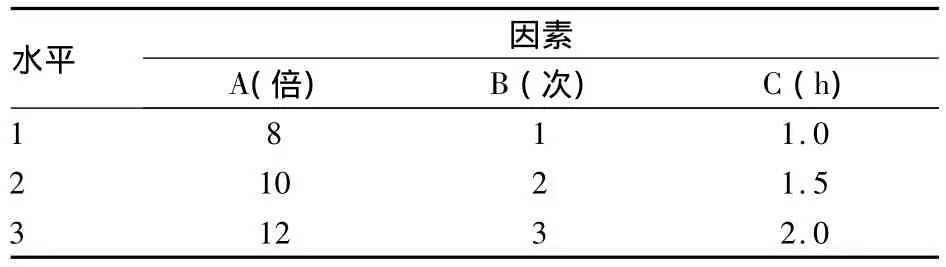
表1 L9(34)水提取工藝因素水平
2.1.2 咖啡酸含量測定 ①色譜條件:Luna C18色譜柱,以甲醇∶0.1%磷酸水溶液(30∶70)為流動相等度洗脫,流速:1 mL/min。檢測波長為323 nm,柱溫25℃。理論板數按咖啡酸峰計算應不低于3 000。②對照品溶液的制備:精密稱取于105℃干燥至恒重的咖啡酸5.06 mg置25 mL量瓶中,加甲醇適量,超聲溶解,放冷并稀釋,得濃度為202.4 μg/mL的咖啡酸對照品儲備液。③供試品溶液的制備:精密稱取相當于蒲公英原藥材0.75 g的藥材干膏粉,移至50 mL錐形瓶中,精密加5%甲酸的甲醇溶液10 mL,密塞,搖勻,稱重,超聲處理(功率250 W,頻率40 kHz)30 min,取出,放冷,稱重,用5%甲酸的甲醇溶液補足失重,搖勻,離心(3 300 r/min,8 min),取上清液,0.22 μm 微孔濾膜濾過,備用。④測定:分別精密吸取對照品溶液與供試品溶液各10 μL,注入液相色譜儀測定。⑤線性關系考察:分別精密量取上述對照品溶液 0.5、1.0、2.0、3.0、4.0、6.0 mL,加甲醇,得濃度為 10.12、20.24、40.48、60.72、80.96、121.44、202.40 μg/mL 的對照品溶液。精密吸取10 μL注入液相色譜儀中測定峰面積,以咖啡酸對照品的峰面積(Y)對進樣量(X)進行線性回歸,得到回歸方程:Y=62 305X-37 581,r=0.999 9。咖啡酸在 10.12 ~202.40 μg 范圍內與色譜峰面積呈良好的線性關系。
2.2 統計學方法 采用SPSS16.0統計軟件,計量數據用ˉx±s表示,數據比較采用方差分析。α=0.05。
2.3 化瘀無糖顆粒提取的正交試驗結果 見表2、3。通過對正交試驗結果直觀分析,各因素影響次序為B(煎煮次數)>C(煎煮時間)>A(加水量)。方差分析表明,B因素對綜合評分有顯著性影響,結合K值分析,確定最佳提取工藝為 A2B3C3,即加入10倍量的水,煎煮3次、2 h/次。
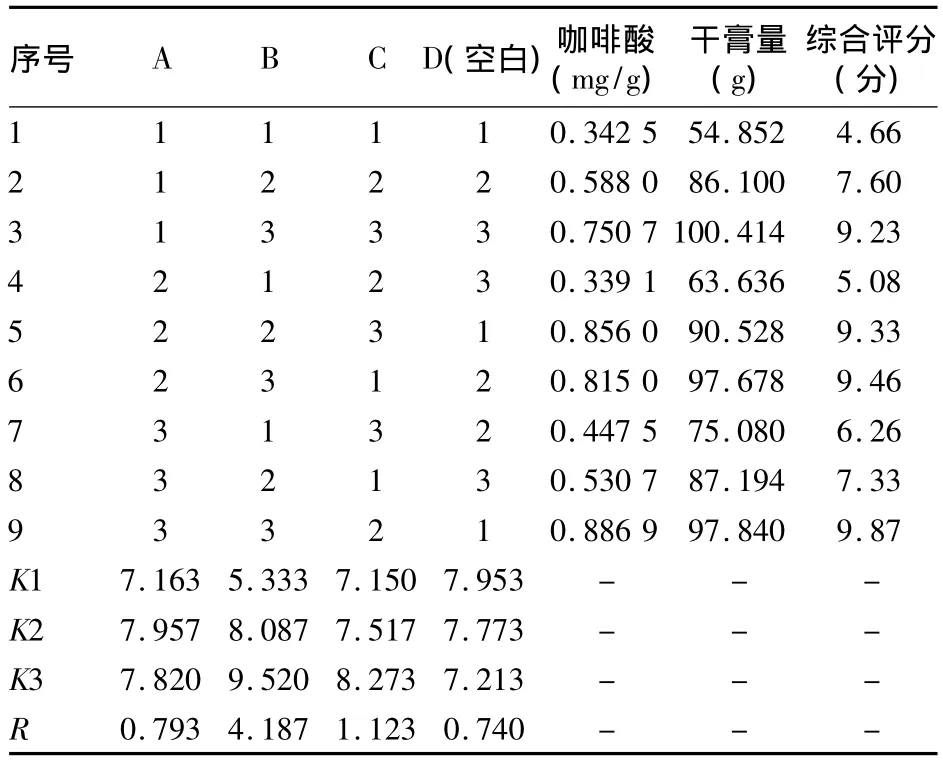
表2 化瘀無糖顆粒提取工藝正交試驗結果
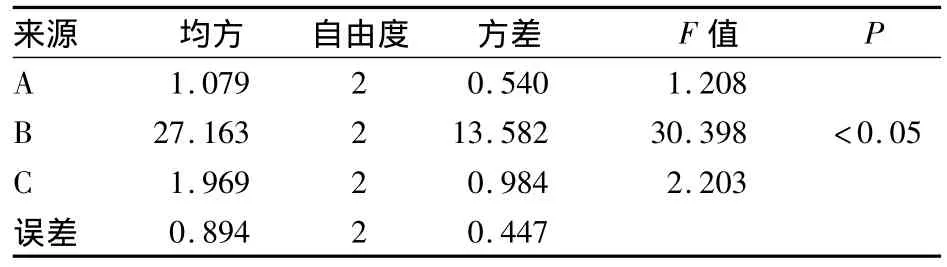
表3 綜合評分方差分析表
2.4 化瘀無糖顆粒提取工藝驗證試驗結果 稱取處方量藥材3份,按上述制備工藝進行驗證,結果與正交實驗結果相符(見表4),說明本提取工藝穩定可行。

表4 化瘀無糖顆粒提取工藝驗證試驗結果
2.5 顆粒成型工藝的篩選
2.5.1 浸膏粉的制備 稱取處方量藥材,按優化所得工藝煎煮提取,合并提取液,濾過,濾液減壓濃縮至相對密度為1.30~1.35的清膏,60℃真空干燥得干浸膏,粉碎,過100目篩,加入人參、水蛭超微粉,混勻,備用。
2.5.2 樣品的制備 將上述浸膏粉與糊精、阿斯巴甜混合均勻,干法[3,4]制粒(設備參數:送料變頻器速率22.0、壓片變頻器轉速20.5、制粒變頻器速率21.0)、整粒、分裝即得。
2.5.3 輔料用量的選擇 選用糊精[5,6]作為賦型劑。取上述干浸膏粉分別加入5%、10%、15%糊精與不加糊精的干浸膏粉分別干法制粒,觀察顆粒的成型性、吸濕性、溶化性[7,8]。
對這3項指標根據其對顆粒劑質量評價的作用綜合評分(權重系數分別為 0.5、0.25、0.25),總分為10分。詳見表5。由表5可知10%糊精為最佳用量。
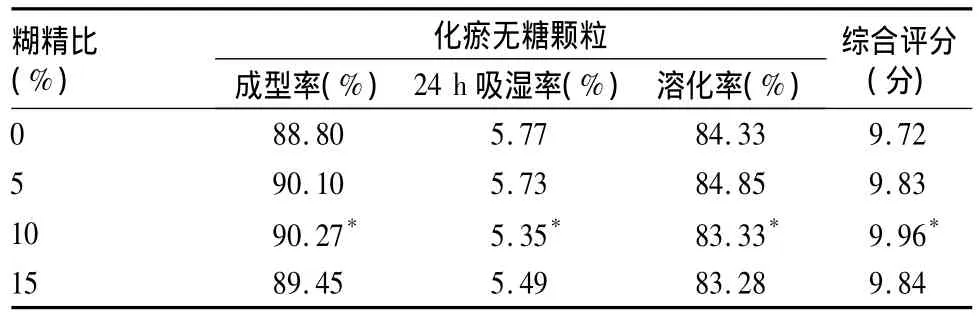
表5 不同比例糊精制化瘀無糖顆粒質量評價情況
2.5.4 顆粒流動性的測定 用休止角[8,9]來衡量,采用固定漏斗法[10],即將3只漏斗串聯并固定于水平放置的坐標紙上適宜高度處,使漏斗下口距坐標紙的距離為H,小心地將樣品沿漏斗壁倒入最上面的漏斗中,直到最下面漏斗藥粉的圓錐體尖端接觸到漏斗口為止,此時由坐標紙測出圓錐體底部的直徑R,計算出休止角(tgα=2H/R)。每個批次測定3次,求其均值。予5批樣品測定,休止角均值分別為28.07°、28.39°、28.72°、28.72°、28.39°,相對標準偏差(RSD)為0.96%,表明顆粒流動性均較好。
2.6 矯味劑用量的篩選 處方中含水蛭原粉,具腥味,且提取物有一定的苦味,故選用阿斯巴甜[11,12]加以矯味,比較阿斯巴甜用量分別為粉末總量0.1%、0.2%、0.3%、0.4%、0.5%時的口感,結果表明阿斯巴甜用量為0.3%時甜度適宜,口感佳。
3 討論
化瘀顆粒為臨床經驗方,功效扶正益氣,化瘀去毒。腫瘤術后及放化療后,機體元氣大傷,五臟俱虛,方中人參苦甘而溫,大補元氣、補脾益肺;蒲公英清熱解毒、消腫散結,可清尚存瘤毒;佐以水蛭等逐瘀行氣、軟堅散結之品,可消瘤毒所致氣血瘀滯,諸藥合用共奏扶正祛邪之效。用于各種腫瘤防治及放療化療、腫瘤術后的輔助治療,臨床應用多年療效確切。
原化瘀顆粒以蔗糖為輔料,易潮解變質,不宜久存;且蔗糖具有較高生物活性,能誘發胃炎、齲齒、糖尿病及肥胖,不宜用于年老及禁糖患者。本試驗在原制劑基礎上進行劑型改革,研制出化瘀無糖顆粒劑型,從而提高了藥物穩定性,保證了臨床療效;改為無糖劑型后可減少輔料用量,便于攜帶和服用;擴大了適應證,解除了糖尿病患者和糖耐量異常的人群的用藥限制[14]。
本試驗旨在研制其無糖劑型,以擴大適應證,故未改變提取溶媒。因其君藥人參以原粉入藥,臣藥中以蒲公英為主藥,故選擇咖啡酸作為含量測定指標結合干膏量的綜合評分篩選提取工藝。根據本試驗結果化瘀無糖顆粒最佳提取工藝條件為A2B3C3,即加10倍量水,煎煮3次,每次2 h。
干法制粒具有輔料用量少、成本低、工藝簡單、周期短、污染小等優點,適用于中藥顆粒劑及片劑的制備,試驗中發現干浸膏粉直接制顆粒成型性尚可,但吸濕性較強易粘連軋輪,故需要加入輔料;在輔料選擇方面,考慮到微晶纖維素水溶性較差、乳糖成本太高、淀粉用量大,故我們選用糊精作為賦型劑。
通過對本顆粒成型工藝的篩選發現,糊精用量為干浸膏量的10%時顆粒的成型性、吸濕性、溶化性均優于其他比例;阿斯巴甜用量為粉末總量的0.3%時甜度適宜。
總之,工藝驗證試驗表明加10倍量水煎煮3次,每次2 h;糊精用量為干浸膏量的10%,阿斯巴甜用量為粉末總量的0.3%,采用干法制粒工藝制得的化瘀無糖顆粒各質量指標均符合《中國藥典》2010年版規定。因此,采用本試驗篩選所得工藝可成功制備化瘀無糖顆粒,且該制備工藝穩定可行。
[1]謝冬梅,劉叢彬.桂枝茯苓混懸型無糖顆粒劑的制備工藝研究[J].安徽醫藥,2005,9(5):328-329.
[2]杜樹山,張錚,沈圣民,等.解毒利尿顆粒水提工藝的正交實驗法優選[J].時珍國醫國藥,2010,21(10):2596-2597.
[3]張毓,鐘曉明.中藥顆粒劑成型工藝的研究進展[J].海峽藥學,2010,22(1):27-28.
[4]魏筱華,賴伊姍,黃愷,等.雙黃連泡騰片干法制粒工藝研究[J].中國實驗方劑學雜志,2010,16(11):1-5.
[5]田原,樊暉,鄂蘊娟.正交設計法優選活血益智片提取工藝實驗研究[J].中華中醫藥學刊,2010,28(11):2318-2319.
[6]李杜軍,劉明生,張鵬威,等.天杞顆粒的制備工藝[J].醫藥導報,2011,30(4):505-507.
[7]張沂,凌云,王強.無糖型咽炎顆粒成型工藝的研究[J].解放軍藥學學報,2010,26(2):123-124.
[8]張亞輝,顧政一,邢建國,等.蒿藍無糖感冒顆粒成型工藝研究[J].時珍國醫國藥,2010,21(10):2597-2598.
[9]宋娟娜,孫文基,王榮,等.清口顆粒無糖型顆粒劑成型工藝的研究[J].第四軍醫大學學報,2008,29(14):1334-1336.
[10]范碧亭,張兆旺,施順清.中藥藥劑學[M].上海:上海科學技術出版社,1997:554.
[11]袁羿,馬龍,徐芳,等.瑣瑣葡萄多糖顆粒制備工藝的研究[J].中成藥,2011,33(1):153-156.
[12]唐建華.新型矯味劑在藥物口服制劑中的應用[J].中國藥業,2007,16(18):59-60.
[13]田旭,王鵬,詹妮,等.抗癌中草藥有效成分的研究進展[J].特產研究,2010,49(3):73-76.
[14]馬家驊,楊明,譚玉婷,等.中藥無糖顆粒劑的研究概況[J].中國藥業,2006,15(5):47-48.
猜你喜歡
中國特種設備安全(2022年5期)2022-08-26 09:19:32
礦產綜合利用(2020年1期)2020-07-24 08:50:40
山東冶金(2019年6期)2020-01-06 07:45:54
收藏界(2019年2期)2019-10-12 08:26:06
世界農藥(2019年2期)2019-07-13 05:55:12
世界農藥(2019年2期)2019-07-13 05:55:10
模具制造(2019年3期)2019-06-06 02:11:00
山東工業技術(2016年15期)2016-12-01 05:30:59
銅業工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
