分散液液微萃取分光光度法測(cè)定食品中汞含量
2012-10-25 01:26:32王俊平李家樂(lè)方國(guó)臻李春江
食品工業(yè)科技 2012年16期
王俊平,李家樂(lè),王 碩,方國(guó)臻,李春江
(天津科技大學(xué),食品營(yíng)養(yǎng)與安全教育部重點(diǎn)實(shí)驗(yàn)室,天津 300457)
分散液液微萃取分光光度法測(cè)定食品中汞含量
王俊平,李家樂(lè),王 碩*,方國(guó)臻,李春江
(天津科技大學(xué),食品營(yíng)養(yǎng)與安全教育部重點(diǎn)實(shí)驗(yàn)室,天津 300457)
建立了分散液液微萃取分光光度法測(cè)定食品中汞含量的方法。使用雙硫腙、三氯甲烷、乙醇分別作為絡(luò)合劑、萃取溶劑、分散溶劑,研究了萃取溶劑和分散溶劑體積、pH、絡(luò)合劑濃度、萃取時(shí)間等影響因素。在最佳條件下,方法的線性范圍為10~200ng·mL-1,相關(guān)系數(shù)(r)為0.9966,檢出限為3ng·mL-1,對(duì)100ng·mL-1汞離子進(jìn)行7次平行測(cè)定的相對(duì)標(biāo)準(zhǔn)偏差為4.3%。該方法可應(yīng)用于魚(yú)和茶葉中汞含量的測(cè)定。
分散液液微萃取,汞,雙硫腙,紫外可見(jiàn)分光光度法
食品中汞含量的測(cè)定是食品污染物監(jiān)測(cè)的重要項(xiàng)目。上世紀(jì)50年代至70年代,通過(guò)日本的“水俁病”事件,人們開(kāi)始認(rèn)識(shí)到汞的危害[1]。金屬汞及甲基汞對(duì)人體健康的危害主要有神經(jīng)毒性、腎臟毒性、免疫毒性、生殖毒性、胚胎發(fā)育毒性等[2]。傳統(tǒng)的液液萃取分光光度法測(cè)定汞含量的方法具有操作繁瑣、有機(jī)溶劑消耗量大、易造成環(huán)境污染等不足。Assadi等[3]提出了分散液液微萃取技術(shù)(Dispersive liquid-liquid microextraction,DLLME)。該技術(shù)具有速度快、操作簡(jiǎn)便、成本低、回收率高、富集系數(shù)高、環(huán)境友好等優(yōu)點(diǎn)。DLLME技術(shù)與電熱原子吸收法[4-5]、火焰原子吸收法[6-7]、電感耦合等離子體發(fā)射光譜法[8-9]聯(lián)用,實(shí)現(xiàn)了對(duì)多種金屬離子的富集測(cè)定。這些方法雖然十分靈敏,但儀器購(gòu)置費(fèi)用和維護(hù)成本高,需要專業(yè)技術(shù)人員,不適用于日常監(jiān)控。紫外可見(jiàn)分光光度法具有操作簡(jiǎn)便、成本低等優(yōu)點(diǎn)。Gharehbaghi等[10]和Tabrizi等[11]將DLLME與紫外可見(jiàn)分光光度法結(jié)合,分別實(shí)現(xiàn)了對(duì)鈷和鐵的測(cè)定。本實(shí)驗(yàn)建立了分散液液微萃取分光光度法測(cè)定食品中汞含量的方法。
1 材料與方法
1.1 材料與儀器
GBW08617汞單元素溶液標(biāo)準(zhǔn)物質(zhì) 中國(guó)標(biāo)準(zhǔn)物質(zhì)計(jì)量中心,1000μg·mL-1;二苯基硫代卡巴腙(雙硫腙) 天津市光復(fù)精細(xì)化工研究所,分析純;甲醇、乙醇、丙酮、氯化鈉、乙二胺四乙酸二鈉、抗壞血酸天津市北方天醫(yī)化學(xué)試劑廠,分析純;乙腈 天津科銳思精細(xì)化工有限公司,分析純;二氯甲烷、三氯甲烷、四氯甲烷 天津市化學(xué)試劑二廠,分析純;四氯乙烯 天津市威晨化學(xué)試有限公司,分析純;鹽酸、無(wú)水醋酸鈉 天津市化學(xué)試劑一廠,分析純;高氯酸、硝酸 天津市化學(xué)試劑五廠,分析純。
Cary 50紫外可見(jiàn)分光光度計(jì) 澳大利亞Varian公司;MP 220酸度計(jì) 瑞士Mettler Toledo公司;Milli-Q超純水系統(tǒng) 美國(guó)Millipore公司;5804 R離心機(jī) 德國(guó)Eppendorf公司。
1.2 實(shí)驗(yàn)方法
1.2.1 分散液液微萃取過(guò)程 分散液液微萃取分光光度法富集測(cè)定汞離子過(guò)程:向15mL刻度離心管中加入0.75mL濃度為2μg·mL-1的汞離子標(biāo)準(zhǔn)工作液,用pH為2.0的鹽酸溶液將體積調(diào)至10.0mL。然后加入0.6g氯化鈉,輕輕搖晃。氯化鈉溶解后,加入100μL 0.02mol·L-1EDTA-Na2溶液和200μL 1.0mol·L-1抗壞血酸溶液,然后加入45μL 1.0×10-3mol·L-1雙硫腙乙醇溶液,反應(yīng)1min,使其形成汞-雙硫腙絡(luò)合物。用5.0mL注射器將1.8mL乙醇(分散溶劑)和75μL三氯甲烷(萃取溶劑)的混合溶劑注入上述溶液中。然后在3500r/min轉(zhuǎn)速下離心3min。使用注射器移去水層,用100μL微量注射器吸取離心管底部的有機(jī)相三氯甲烷(體積約為50μL)至光程為1cm、容積為500μL的比色皿,使用丙酮將其稀釋至300μL。然后用分光光度計(jì)在波長(zhǎng)495nm處以試劑空白作參比測(cè)定吸光度。試劑空白中不加汞離子標(biāo)準(zhǔn)工作液,其余試劑和操作與標(biāo)準(zhǔn)工作液的處理相同。
1.2.2 標(biāo)準(zhǔn)曲線的繪制 分別移取0.05、0.25、0.5、0.75、1.0mL濃度為2μg·mL-1的汞離子標(biāo)準(zhǔn)工作液于刻度離心管中,用pH為2.0的鹽酸溶液將體積調(diào)至10.0mL。按照1.2.1的方法操作,測(cè)定吸光度后,繪制吸光度-濃度標(biāo)準(zhǔn)曲線并計(jì)算回歸方程。
1.2.3 樣品的消化與測(cè)定 魚(yú)肉取可食部分打成勻漿,茶葉粉碎,保存于塑料瓶中備用。準(zhǔn)確稱取試樣1.00~2.00g于高腳燒杯中,加入10.0mL硝酸和2.0mL高氯酸,蓋上表面皿浸泡過(guò)夜。在電熱板上小火加熱,然后慢慢加大火力至溶液澄清無(wú)色或微帶黃色,使溶液體積蒸發(fā)至1~2mL,冷卻至室溫。用少量水洗滌燒杯壁,用氨水將溶液pH調(diào)至2.0,將全部液體轉(zhuǎn)移至10mL容量瓶,用pH為2.0鹽酸水溶液定容。用10.0mL樣品溶液替代1.2.1中體積調(diào)節(jié)至10.0mL后的汞離子標(biāo)準(zhǔn)工作液,按照1.2.1的方法操作。
2 結(jié)果與討論
2.1 吸收光譜
在400~700nm波長(zhǎng)范圍內(nèi)對(duì)試劑空白和汞-雙硫腙絡(luò)合物的吸收光譜進(jìn)行了測(cè)定。圖1表明,丙酮作為參比溶劑時(shí),試劑空白與絡(luò)合物的最大吸收波長(zhǎng)分別位于600、490nm處,Δλ=110nm,最大吸收波長(zhǎng)差別較大,利于分光光度法測(cè)定。以試劑空白為參比時(shí),絡(luò)合物的最大吸收波長(zhǎng)位于495nm處。本研究選擇495nm作為測(cè)定波長(zhǎng)。
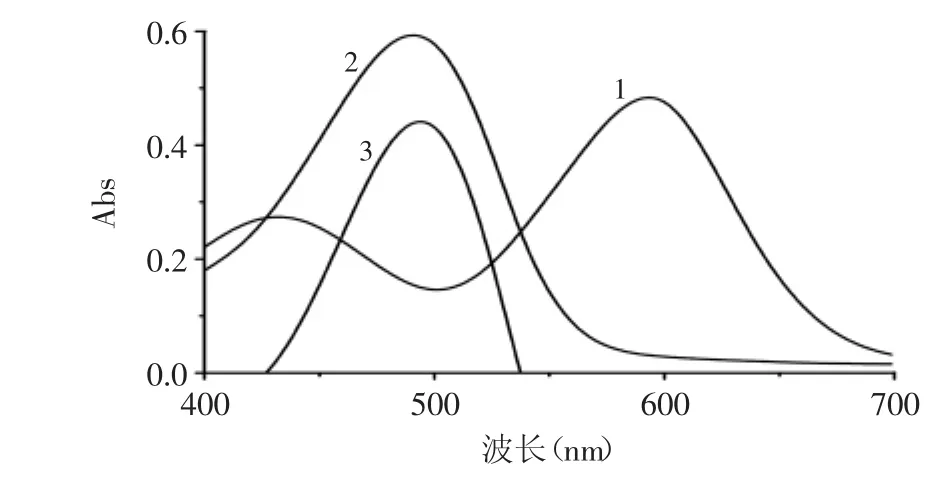
圖1吸收光譜Fig.1 Absorption spectra
2.2 萃取溶劑和分散溶劑的選擇
萃取溶劑的種類是影響分散液液微萃取的重要因素。它通常需要滿足密度比水大、在水中溶解度較小、對(duì)目標(biāo)物有較高的萃取能力三個(gè)條件。分散溶劑要能夠與水和萃取溶劑互溶,它可促使體系形成穩(wěn)定的乳濁液并將萃取溶劑均勻地分散于水中,使萃取溶劑形成微小液滴,增大萃取溶劑與目標(biāo)物的接觸面積。分別使用200μL不同萃取溶劑(二氯甲烷、三氯甲烷、四氯甲烷、四氯乙烯)和1.8mL不同分散溶劑(甲醇、乙醇、丙酮、乙腈)進(jìn)行實(shí)驗(yàn)。圖2表明,在所有組合中,使用三氯甲烷和乙醇分別作為萃取溶劑和分散溶劑時(shí)吸光度最大。因此,本實(shí)驗(yàn)選擇三氯甲烷作為萃取溶劑,乙醇作為分散溶劑。
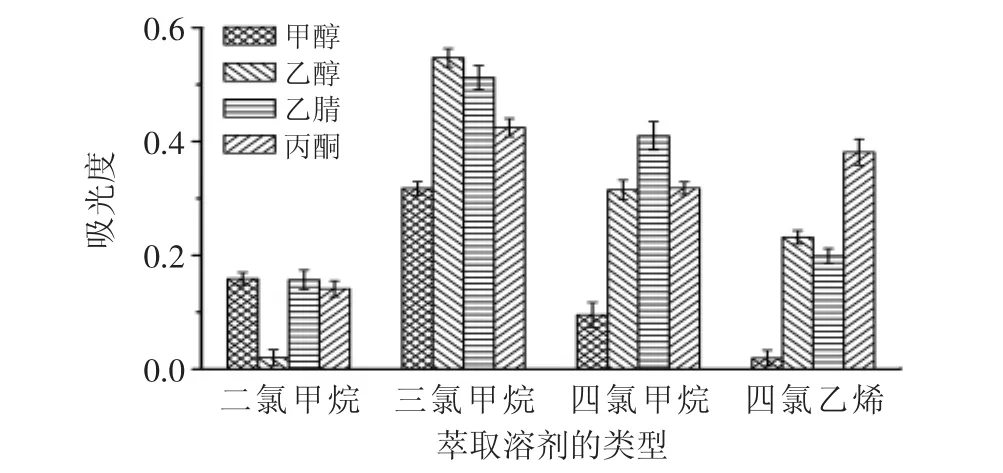
圖2 萃取溶劑和分散溶劑的選擇Fig.2 Selection of the type of extraction and disperser solvent
2.3 萃取溶劑體積對(duì)吸光度的影響
研究了不同體積的三氯甲烷對(duì)吸光度的影響。隨著三氯甲烷體積的增加,吸光度逐漸增大。當(dāng)三氯甲烷用量為75μL時(shí),吸光度達(dá)到最大。其用量為100、125μL時(shí)吸光度并沒(méi)有繼續(xù)增大。本實(shí)驗(yàn)選擇三氯甲烷的用量為75μL。
2.4 分散溶劑體積對(duì)吸光度的影響
研究了不同體積乙醇對(duì)吸光度的影響。圖3表明,隨著乙醇用量從0.3mL增加至1.8mL,吸光度由0.12增加到0.54。當(dāng)分散溶劑用量較少時(shí),體系形成的乳濁液不穩(wěn)定,萃取溶劑在水中形成的液滴較大并且快速沉降到離心管底部,導(dǎo)致萃取溶劑液滴與目標(biāo)物的總接觸面積較小且接觸時(shí)間較短,所以萃取效率低。當(dāng)乙醇體積為1.8mL時(shí)吸光度最大,隨著其體積繼續(xù)增加,吸光度又緩慢減小。這是因?yàn)榉稚⑷軇w積過(guò)大時(shí),絡(luò)合物在水相中的溶解量也隨之增加,故引起吸光度的下降。本實(shí)驗(yàn)選擇乙醇的用量為1.8mL。
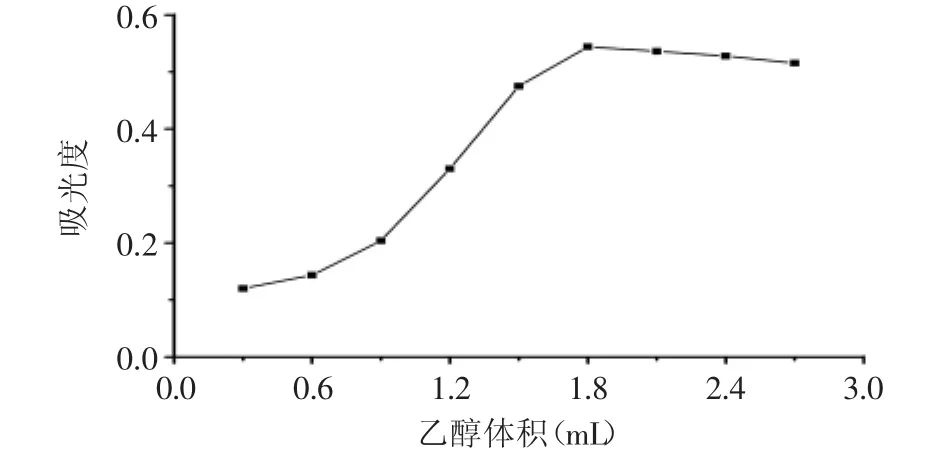
圖3 乙醇體積對(duì)吸光度影響Fig.3 Effect of volume of ethanol on the absorption
2.5 酸度對(duì)吸光度的影響
樣品溶液酸度不僅影響汞-雙硫腙絡(luò)合物的形成、配位數(shù)、穩(wěn)定性,而且影響后續(xù)的分散液液微萃取過(guò)程。在pH為0.5~3.5范圍內(nèi)研究了酸度對(duì)吸光度的影響。圖4中的結(jié)果表明,pH控制在2.0時(shí)吸光度最大。
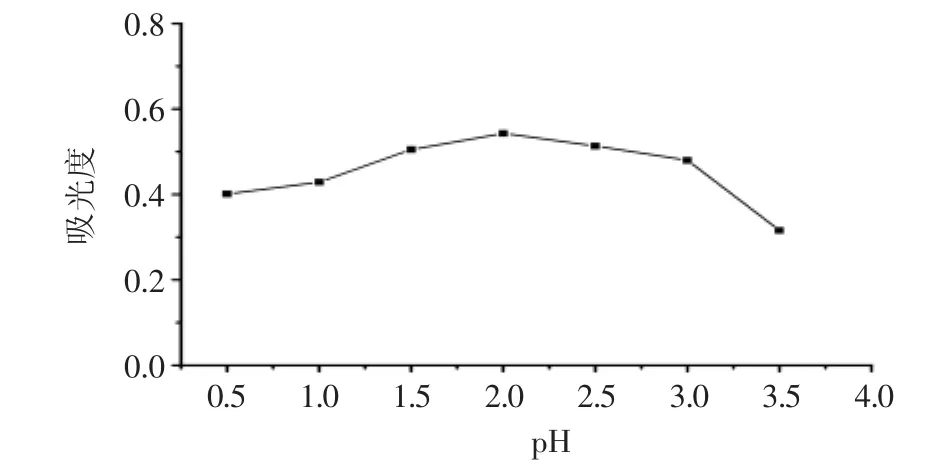
圖4pH對(duì)吸光度的影響Fig.4 Effect of pH on the absorption
2.6 絡(luò)合劑用量對(duì)吸光度的影響
在本實(shí)驗(yàn)中,絡(luò)合劑的作用體現(xiàn)在兩方面:一方面,絡(luò)合劑可以與汞離子發(fā)生絡(luò)合反應(yīng)生成絡(luò)合物,疏水的絡(luò)合物在分散液液微萃取過(guò)程中易于被萃取溶劑萃取;另一方面,絡(luò)合劑作為金屬離子顯色劑,是分光光度法定量測(cè)定的基礎(chǔ)。因此,絡(luò)合劑的濃度直接影響絡(luò)合物的形成、絡(luò)合物的萃取以及分光光度法的信號(hào)強(qiáng)度。研究了不同濃度雙硫腙對(duì)于汞離子測(cè)定的影響。結(jié)果表明,隨著雙硫腙濃度的增大,吸光度隨之增大,當(dāng)雙硫腙濃度為4.5μmol·L-1時(shí)吸光度達(dá)到最大,然后趨于穩(wěn)定。本實(shí)驗(yàn)選擇雙硫腙濃度為4.5μmol·L-1。
2.7 萃取時(shí)間對(duì)吸光度的影響
在分散液液微萃取技術(shù)中,萃取時(shí)間被定義為溶劑注射與離心分離兩個(gè)操作步驟之間的時(shí)間間隔。研究了不同萃取時(shí)間對(duì)吸光度的影響。由圖5可知,隨著萃取時(shí)間由1min增加到5min,吸光度緩慢下降;在萃取時(shí)間由5min繼續(xù)增加到15min過(guò)程中,吸光度下降的速度加快。以往的研究認(rèn)為,在分散液液微萃取過(guò)程中,萃取溶劑被均勻分散為極小的液滴,萃取溶劑和水相之間的接觸表面積趨于無(wú)限大,目標(biāo)物從水相遷移至萃取溶劑的速度非常快,所以能夠迅速達(dá)到萃取平衡態(tài),因而萃取時(shí)間對(duì)分析信號(hào)不會(huì)產(chǎn)生明顯的影響。但在本實(shí)驗(yàn)中,因?yàn)殡p硫腙自身穩(wěn)定性較差,易被氧化分解,導(dǎo)致吸光度隨萃取時(shí)間延長(zhǎng)而下降。為了避免雙硫腙的氧化分解,向體系中加入200μL 1.0mol·L-1的抗壞血酸溶液,體系吸光度雖然也隨著萃取時(shí)間的延長(zhǎng)有所下降,但下降速度明顯減緩。為了控制吸光度的穩(wěn)定,向體系中加入200μL 1.0mol·L-1的抗壞血酸溶液并把萃取時(shí)間控制在5min之內(nèi)。

圖5 萃取時(shí)間對(duì)吸光度的影響Fig.5 Effect of extraction time on the absorption
2.8 離心速率對(duì)吸光度的影響
溶劑注射之后體系形成“水/分散溶劑/萃取溶劑”乳濁液三元體系,疏水的萃取溶劑微小液滴懸浮于水中或者吸附在離心管內(nèi)壁,需要通過(guò)離心實(shí)現(xiàn)分離。在不同離心速率(0~4500r/min)條件下離心3min。結(jié)果表明,隨著離心速率的加快,吸光度增大。當(dāng)離心速率達(dá)到3000r/min時(shí),吸光度達(dá)到最大。繼續(xù)增大離心速率,吸光度保持恒定。離心條件設(shè)定為轉(zhuǎn)速3500r/min,時(shí)間3min。
2.9 鹽效應(yīng)
離子強(qiáng)度是影響傳統(tǒng)液液萃取的重要因素。一般來(lái)講,隨著離子強(qiáng)度的增加,目標(biāo)物和有機(jī)萃取溶劑在水相中的溶解度減小,利于提高回收率。在氯化鈉濃度為0%~8%(w/v)范圍內(nèi)研究了離子強(qiáng)度對(duì)吸光度的影響。結(jié)果表明,吸光度隨鹽濃度的增大而增大,當(dāng)鹽濃度達(dá)到5%時(shí)吸光度達(dá)到最大,隨后基本保持恒定。為了穩(wěn)定體系的離子強(qiáng)度,選擇氯化鈉的加入量為6%(w/v)。
2.10 稀釋溶劑
離心之后,離心管底部有機(jī)相三氯甲烷的體積僅有50μL,無(wú)法直接在比色皿中用分光光度法進(jìn)行測(cè)定,需要選擇合適的溶劑對(duì)有機(jī)相進(jìn)行稀釋。考察了甲醇、乙醇、乙腈、丙酮四種常用稀釋溶劑的效果。結(jié)果表明,用丙酮作稀釋溶劑時(shí)吸光度最大。
2.11 干擾離子及干擾消除
在最佳實(shí)驗(yàn)條件下,研究了各種離子對(duì)50ng·mL-1汞離子測(cè)定的干擾。如果共存離子使測(cè)定結(jié)果偏離了±5%,這個(gè)濃度就可以認(rèn)為是該共存離子的最大允許量。由表1可知,對(duì)測(cè)定干擾嚴(yán)重的有Cd2+、Cu2+、Ni2+、Pb2+。使用2×10-4mol·L-1EDTA-Na2作掩蔽劑可有效消除干擾。
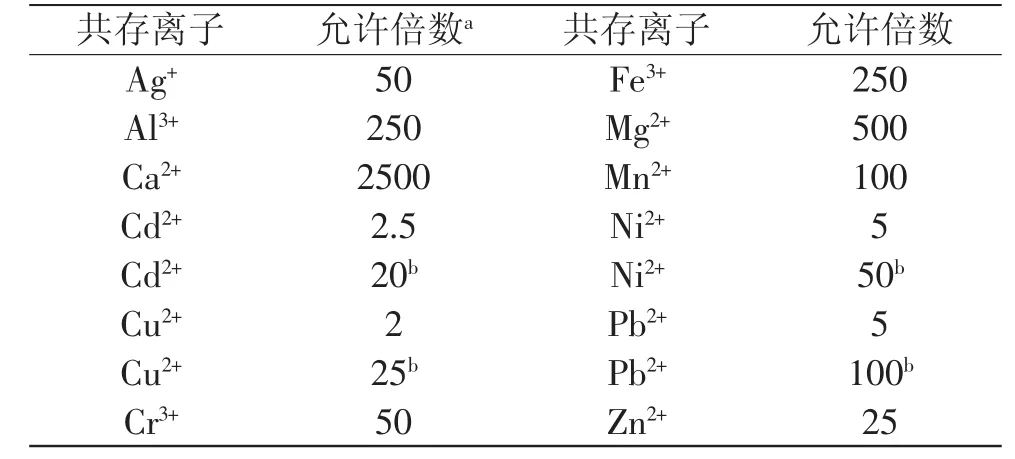
表1 共存離子對(duì)汞測(cè)定的影響Table 1 Effect of foreign ions on the determination of mercury
2.12 分析特征量
汞含量在10~200ng·mL-1范圍內(nèi)符合朗勃比爾定律,回歸方程為A=3.4784C(μg·mL-1)+0.0126,r=0.9966。檢出限為3ng·mL-1(CL=3SB/m,其中CL為檢出限,SB為對(duì)試劑空白進(jìn)行7次測(cè)定所得吸光度的標(biāo)準(zhǔn)偏差,m為標(biāo)準(zhǔn)曲線的斜率)。對(duì)100ng·mL-1的汞離子進(jìn)行7次平行測(cè)定,相對(duì)標(biāo)準(zhǔn)偏差為4.3%。
2.13 方法的應(yīng)用
對(duì)不同樣品進(jìn)行處理和測(cè)定,并進(jìn)行了加標(biāo)回收實(shí)驗(yàn)。結(jié)果見(jiàn)表2,加標(biāo)回收率在96.2%~103.0%之間,表明本方法準(zhǔn)確度較高。
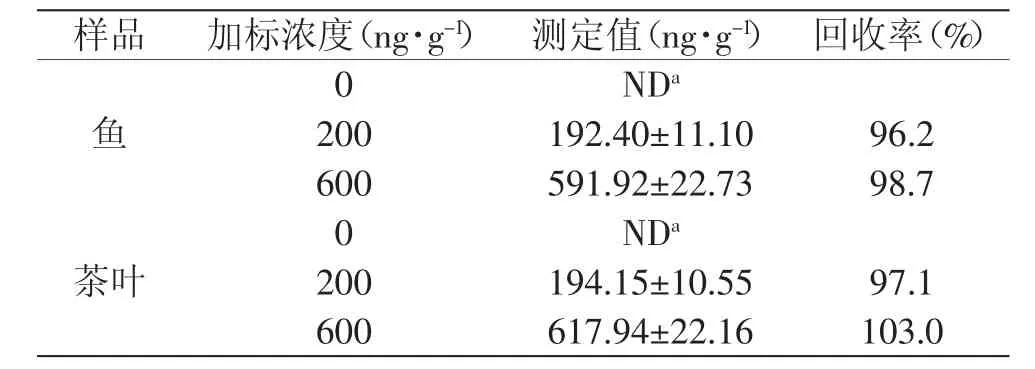
表2 實(shí)際樣品中汞含量測(cè)定Table 2 Determination of mercury in real samples
3 結(jié)論
本實(shí)驗(yàn)將分散液液微萃取技術(shù)與分光光度法相結(jié)合,建立了食品中汞含量的測(cè)定方法。該方法具有簡(jiǎn)便、快速、儀器成本低等優(yōu)點(diǎn)。方法的線性范圍為10~ 200ng·mL-1,檢出限為3ng·mL-1。該方法可應(yīng)用于魚(yú)和茶葉中汞含量的測(cè)定,回收率在96.2%~103.0%之間。
[1]徐蘊(yùn),程欣.環(huán)境汞污染對(duì)人體健康的影響[J].江蘇預(yù)防醫(yī)學(xué),2006,17(3):85-86.
[2]鄭徽,金銀龍.汞的毒性效應(yīng)及作用機(jī)制研究進(jìn)展[J].衛(wèi)生研究,2006,35(5):663-666.
[3]Rezaee M,Assadi Y,Milani Hosseini MR,et al.Determination of organic compounds in water using dispersive liquid-liquid microextraction[J].Journal of Chromatography A,2006,1116(1-2):1-9.
[4]Naseri MT,Milani Hosseini MR,Assadi Y,et al.Rapid determination of lead in water samples by dispersive liquid-liquid microextraction coupled with electrot hermal atomic absorption spectrometry[J].Talanta,2008,75(1):56-62.
[5]Kagaya S,Takata D,Yoshimori T,et al.A sensitive and selective method for determination of gold(III)based on electrothermal atomic absorption spectrometry in combination with dispersive liquid-liquid microextraction using dicyclohexylamine[J].Talanta,2010,80(3):1364-1370.
[6]Kokya TA,F(xiàn)arhadi K.Optimization of dispersive liquidliquid microextraction for the selective determination of trace amounts of palladium by flame atomic absorption spectroscopy [J].Journal of Hazardous Materials,2009,169(1-3):726-733.
[7]Naseri MT,Hemmatkhah P,Hosseini MRM,et al.Combination of dispersive liquid-liquid microextraction with flame atomic absorption spectrometry using microsample introduction for determination of lead in water samples[J].Analytica Chimica Acta,2008,610(1):135-141.
[8]Rezaee M,Yamini Y,Khanchi A,et al.A simple and rapid new dispersive liquid-liquid microextraction based on solidification of floating organic drop combined with inductively coupled plasma-optical emission spectrometry for preconcentration and determination of aluminium in water samples[J].Journal of Hazardous Materials,2010,178(1-3):766-770.
[9]Sereshti H,Khojeh V,Samadi S.Optimization of dispersive liquid-liquid microextraction coupled with inductively coupled plasma-optical emission spectrometry with the aid of experimental design for simultaneous determination of heavy metals in natural waters[J].Talanta,2011,83(3):885-890.
[10]Gharehbaghi M,Shemirani F,Baghdadi M.Dispersive liquidliquid microextraction and spectrophotometric determination of cobalt in water samples[J].International Journal of Environmental Analytical Chemistry,2008,88(7):513-523.
[11]Tabrizi AB.Development of a dispersive liquid-liquid microextraction method for iron speciation and determination in different water samples[J].Journal of Hazardous Materials,2010,183(1-3):688-693.
Development of a dispersive liquid-liquid microextraction method for the spectrophotometric determination of mercury in food samples
WANG Jun-ping,LI Jia-le,WANG Shuo*,F(xiàn)ANG Guo-zhen,LI Chun-jiang
(Key Laboratory of Food Nutrition and Safety,Ministry of Education of China,Tianjin University of Science&Technology,Tianjin 300457,China)
Dispersive liquid-liquid microextraction(DLLME)technique combined with ultraviolet-visible spectrophotometry for the preconcentration and determination of mercury in food samples was developed. Dithizone,chloroform and ethanol were used as chelating agent,extraction solvent and disperser solvent,respectively.Some important DLLME parameters such as the volume of extraction and disperser solvent,pH,concentration of chelating agent and extraction time were investigated in detail.Under the optimized conditions,the calibration graph was linear from 10 to 200ng·mL-1with a correlation coefficient of 0.9966.The limit of detection was 3ng·mL-1and the relative standard deviation(n=7,for 100ng·mL-1of mercury)was 4.3%. The method was successfully applied into the determination of trace amounts of mercury in fish and tea leaves.
dispersive liquid-liquid microextraction;mercury;dithizone;ultraviolet-visible spectrophotometry
TS207.5+1
A
1002-0306(2012)16-0057-04
2011-12-30 *通訊聯(lián)系人
王俊平(1969-),男,博士,教授,研究方向:食品安全檢測(cè)。
“863”計(jì)劃項(xiàng)目(2011AA100806)。
