肝豆?fàn)詈俗冃阅XCT和MRI診斷
2012-11-21 02:49:08范輝王文獻(xiàn)孫傳順岳恒志王珂王文獻(xiàn)石拓劉錦濤何艷
中國實(shí)用醫(yī)藥 2012年17期
關(guān)鍵詞:信號(hào)
范輝 王文獻(xiàn) 孫傳順 岳恒志 王珂 王文獻(xiàn) 石拓 劉錦濤 何艷
肝豆?fàn)詈俗冃?hepatolenticular degeneration HLD)最初由Wilson等[1]于1912年作了較全面描述,又稱wilson病。是一種少見的常染色體隱性遺傳銅代謝障礙疾病,其遺傳缺陷是在染色體13的長鏈上,1921年正式命名為肝豆?fàn)钭冃浴T摬∈装l(fā)癥狀多樣化,伴有多臟器損害,早期誤診率很高[2]。近年來隨著CT及MRI技術(shù)的廣泛應(yīng)用,HLD臨床發(fā)現(xiàn)率及早期確診率明顯有所提高,本組總結(jié)36例HLD的CT、MRI和臨床資料,對(duì)有關(guān)問題進(jìn)行探討。
1 資料與方法
36例(HLD)患者中,男20例,女16例,年齡10~38歲,平均14.5歲,陽性家族史21例,36例中,有明確肝炎史26例,肝炎史不祥7例,3例無肝炎史,檢測(cè)血鈣的25例中19例降低,為0.23~1.05 mmol/L,堿性磷酸酶8例升高,達(dá)156~212 U/L。眼科裂隙燈檢查異常。KayserFleischer(K-F)環(huán)陽性36例,血液生化異常,血清銅氧化酶活力下降和血清銅藍(lán)蛋白水平下降,和尿銅含量增高。典型錐體外系神經(jīng)癥狀30例,不典型肝、腎、胃腸道等癥狀6例。影像學(xué)檢查異常36例。其中腦MR檢查30例,腦CT檢查26例。
CT機(jī)使用使用東芝16排螺旋CT機(jī)掃描,以O(shè)M為基線,窗寬90 mm,窗位30 mm,常規(guī)則層厚及層距為8 mm掃描,病灶部位加層厚及層距5 mm薄層掃描。MR檢查用使用Simens 1.0T超導(dǎo)型和Hitachi AipisII0.3T永磁型磁共振成像機(jī),頭部線圈,層厚6~8 mm,間距1~2 mm,SE序列T1WI:TE15 ~26 ms,TR350 ~500 ms;T2WI:TE80 ~120 ms,TR3500~5000 ms,F(xiàn)IR 序列 TR8500 ms,TE120 ms。常規(guī)橫斷面T1WI+T2WI+FIR+矢狀面T1WI。部分加做矢狀面和冠狀面重T2WI。
2 結(jié)果
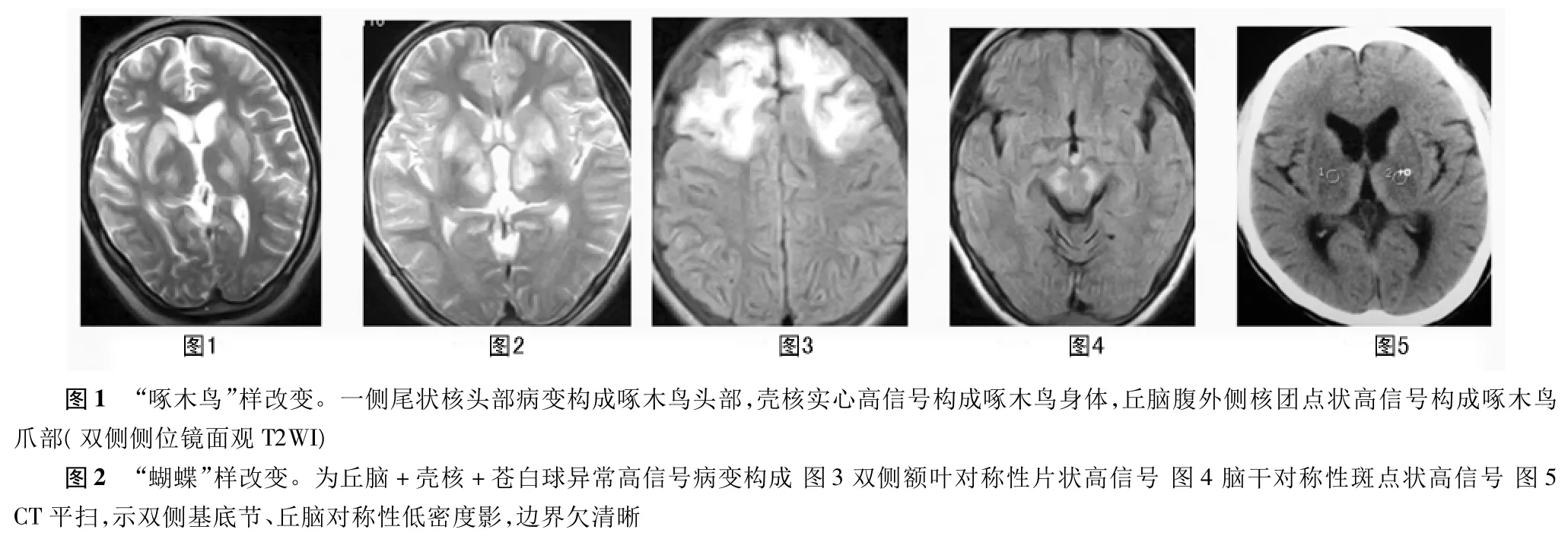
MRI表現(xiàn):30例均可見雙側(cè)基底節(jié)區(qū)對(duì)稱性條狀或新月形異常信號(hào)。其中殼核30例,尾狀核16例,丘腦15例,蒼白球、紅核各3例,黑質(zhì)、大腦腳網(wǎng)狀結(jié)構(gòu)、腦橋橋核、額葉白質(zhì)各2例(圖1~4)。主要呈長T1長T2信號(hào),F(xiàn)IR高信號(hào)。典型者在T2WI及FIR圖像基底節(jié)層面上,依據(jù)受累基底節(jié)神經(jīng)核團(tuán)的不同,分別表現(xiàn)為“啄木鳥”、“八字”、“雙八字”和“展翅蝴蝶”樣改變等征象。啄木鳥樣改變?yōu)閭?cè)位觀鏡面像,由兩側(cè)尾狀核頭部病變構(gòu)成啄木鳥頭部,殼核實(shí)心高信號(hào)或周邊殼樣高信構(gòu)成啄木鳥身體,丘腦腹外側(cè)核團(tuán)呈點(diǎn)狀高信號(hào)構(gòu)成啄木鳥瓜部,雙側(cè)則形成啄木鳥側(cè)面鏡像(圖1)。T1WI上上述影像表現(xiàn)為長T1較低信號(hào)。必須注意正常人尾核頭、殼核在T2圖像上也呈稍高信號(hào),但顯著輕微,絕不亮白致密,更無明暗不均,且無丘腦病變,可資鑒別。丘腦病變?nèi)比缯撸瑒t呈“缺瓜啄木鳥”樣改變,“八字樣”改變?yōu)閮H有殼核病變者,“雙八字”樣變?yōu)闅ず?丘腦病變,如丘腦病變范圍較大,加上豆?fàn)詈瞬∽儯瑒t呈“展翅蝴蝶”樣改變(圖2)。上述各種腦部MR改變均有殼核受累。26例HLD患者CT檢查顯示基底節(jié)區(qū)對(duì)稱性的低密度影,大小、范圍、形態(tài)、密度近似(圖5),可位于豆?fàn)詈恕⑽矤詈恕⑶鹉X區(qū)和額葉等部位,其中5例CT僅發(fā)現(xiàn)基底節(jié)區(qū)病灶,MR側(cè)發(fā)現(xiàn)大腦腳、橋核多處病灶。15例患者輕度或中度腦萎縮,表現(xiàn)為腦室擴(kuò)大及不同程度的腦溝裂增寬等腦萎縮表現(xiàn)。
3 討論
HLD 因其病理特征為肝硬化和腦基底節(jié)區(qū)的豆?fàn)詈俗冃远妹1静∈浅H旧w隱性遺傳性銅代謝障礙疾病,銅藍(lán)蛋白(CP)合成障礙是本病最基本的遺傳缺陷,患者血清銅降低,由于游離銅離子不能與銅蛋白結(jié)合,過量銅沉積在肝、腦、腎和角膜組織而致病,整個(gè)神經(jīng)系統(tǒng)均可受累,主要累及組成錐體外系的基底節(jié)神經(jīng)核團(tuán),包括豆?fàn)詈恕⑽矤詈恕⑵翣詈艘约板F體系功能有關(guān)的丘腦底核、黑質(zhì)和紅核。其中尤以豆?fàn)詈藘?nèi)的殼核最為明顯,其次為蒼白球及尾狀核。殼核最早發(fā)生變性、萎縮,色素沉著加深,類似變化還可出現(xiàn)在小腦齒狀核及大腦皮質(zhì)等部位。腦MRI表現(xiàn)可反映病理變化,最多見和最具特征性的征象是雙側(cè)豆?fàn)詈藢?duì)稱性異常信號(hào),同時(shí)MRI表現(xiàn)能反映本病臨床嚴(yán)重程度,有殼核異常信號(hào)者最重,其次為蒼白球異常信號(hào)灶,僅有腦萎縮者最輕,殼核受累常出現(xiàn)對(duì)稱性條形或新月形異常信號(hào)區(qū)、形如“八字”或“展翅蝴蝶”這與其解剖結(jié)構(gòu)是一致的。
MRI因?qū)π盘?hào)變化的敏感及沒有骨偽影,對(duì)肝豆?fàn)詈俗冃阅X部病變檢出明顯示優(yōu)于CT[1]。肝豆核變性腦部病變的分布頻率有多種的報(bào)道,Roh等[2]報(bào)道25例治療前病例,病變分布為雙側(cè)丘腦92%,殼核68%,蒼白球40%,尾狀核20%,小腦16%,腦白質(zhì)4%;King等[3]報(bào)道22例分布為丘腦50%,殼核40%,蒼白球41%,中腦75%,橋腦82%,小腦50%,本組30例患者M(jìn)R檢查均有腦部信號(hào)異常,分布為殼核30例,尾狀核16例,丘腦15例,蒼白球、紅核各3例,黑質(zhì)、大腦腳網(wǎng)狀結(jié)構(gòu)、腦橋橋核、額葉白質(zhì)各2例,因各家報(bào)道所入選的病例數(shù)不一,所有機(jī)器不同,治療與治療時(shí)間長短、病變分布頻率相差較大,幾乎所有區(qū)域的灰白質(zhì)都可受累,病變分布頻率最高的是豆?fàn)詈?36% ~72%)[1-4],其次是腦干[1-4],在腦干,病灶往往是對(duì)稱分布在大腦腳、中腦被蓋、黑質(zhì)紅核及導(dǎo)水管四周灰質(zhì)[2]。在丘腦病變中,異常信號(hào)往往是對(duì)稱的。Haan等[5]將病變信號(hào)分為3種,第一種是T2WI高信號(hào)為主,第二種是T1WI高信號(hào)為主,第三種是T2WI低信號(hào)。第一種病變信號(hào)為最常見,可發(fā)生在基底節(jié)、丘腦及及下丘腦區(qū)域。現(xiàn)在普遍認(rèn)為在灰質(zhì)的T2WI高信號(hào)是由于水腫、腦質(zhì)增生、神經(jīng)元壞死及囊樣變性所致。在白質(zhì)的T2WI高信號(hào)是由于脫髓鞘、軟化、海綿樣變性及空洞形成。T2WI高信與銅離子的順磁性無關(guān),可能因?yàn)殂~離子以細(xì)胞內(nèi)形式聚集[5]。第二種病變信號(hào)主要發(fā)生在蒼白球[6],本組有4例尾狀核及豆?fàn)詈薚2WI高信號(hào),第三種異常信號(hào)主要發(fā)生在基底節(jié)[1、3],本組有2例下丘腦低信號(hào),可能患者在未經(jīng)治療或治療較差時(shí),銅在腦組織中的聚集量逐漸增多,其順磁作用明顯超過了水腫和膠質(zhì)化所造成T2WI高信號(hào)。腦萎縮是本癥較常見的腦部異常,彌漫性腦萎縮較局限性腦萎縮更常見。Grimm[7]文獻(xiàn)報(bào)道肝豆?fàn)詈俗冃曰颊叩哪X萎縮發(fā)生率為68%,本組腦萎縮的發(fā)生率較低(42.0%),可能與本組病例年齡較小和病程較短有關(guān)。現(xiàn)在普遍認(rèn)為腦萎縮與銅的毒性有關(guān)。由于肝豆?fàn)詈俗冃曰颊叩倪z傳缺陷,銅不能排入膽汁而在肝細(xì)胞中聚集起來并對(duì)肝細(xì)胞造成損傷,壞死的肝細(xì)胞再釋放銅損傷其他臟器,如腦、腎臟等[8,9]。因此肝臟受損往往是肝豆?fàn)詈俗冃詢和畛鹾妥铒@著的表現(xiàn)。
總之,MRI作為一種無創(chuàng)傷性檢查手段,其典型表現(xiàn)結(jié)合CT及臨床表現(xiàn),角膜K-F環(huán),血、尿銅及銅藍(lán)蛋白等生化指標(biāo),對(duì)肝豆?fàn)詈俗冃缘脑\斷及預(yù)后具有重要意義。鑒別診斷主要包括中毒性腦病和缺氧性腦病等。
[1]Magalhaes ACA,Garamelli P,Menezes JR,et al.Wilson's disease:MRI with clinical correlation.Neuroradiology,1994,36:97.
[2]King D,Walshe M,Kendall E,et al.Cranial MR imgaging in wilson's aisease.AJR,1996,167:1579.
[3]劉祥普.肝豆?fàn)詈俗冃缘呐R床影像表現(xiàn)及其診斷.醫(yī)藥論壇雜志,2011,32(7):186-187.
[4]盧虹.肝豆?fàn)詈俗冃缘腃T診斷與臨床分析.醫(yī)學(xué)綜述,2011,(11):154-155.
[5]周豐,朱彩云,廖祥福.兒童肝豆?fàn)詈俗冃哉`診20例分析.中國誤診學(xué)雜志,2010,10(10):132.
[6]黃躍金,鄔至平,蔡學(xué)禮.肝豆?fàn)詈俗冃?8例臨床特點(diǎn)與CT.MRI分析.中國實(shí)用神經(jīng)疾病雜志,2006,9(3):131-132.
[7]高文清,孔玲玲,劉鵬程,等.肝豆?fàn)詈俗冃缘挠跋駥W(xué)表現(xiàn)及成像相關(guān)因素探討.中華放射學(xué)雜志,2002,36(5):402.
[8]盧高峰,唐芙愛.肝豆?fàn)詈俗冃?6例臨床分析.中國實(shí)用神經(jīng)疾病雜志,2011,14(15):39-40.
[9]梁秀軍,展曉梅.肝豆?fàn)詈俗冃?2例誤診分析.中國當(dāng)代醫(yī)藥,2011,18(13):135-137.
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評(píng)價(jià)·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(hào)(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(hào)(2018年2期)2018-04-18 12:18:10
鐵道通信信號(hào)(2016年11期)2016-06-01 12:11:32
鑿巖機(jī)械氣動(dòng)工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
