CaO改性對CuZnAlZr催化劑在合成氣制低碳醇中性能的影響
2012-12-11 09:11:48朱秋鋒張榮俊賀德華
物理化學(xué)學(xué)報 2012年6期
朱秋鋒 張榮俊 賀德華
(清華大學(xué)化學(xué)系,有機光電子與分子工程教育部重點實驗室,分子催化與定向轉(zhuǎn)化研究室,北京100084)
CaO改性對CuZnAlZr催化劑在合成氣制低碳醇中性能的影響
朱秋鋒 張榮俊 賀德華*
(清華大學(xué)化學(xué)系,有機光電子與分子工程教育部重點實驗室,分子催化與定向轉(zhuǎn)化研究室,北京100084)
采用機械混合法和共沉淀法制備了CaO改性CuZnAlZr氧化物催化劑,在高壓微型固定床反應(yīng)器上考察了其在合成氣制低碳醇反應(yīng)中的催化性能,利用粉末X射線衍射(XRD)、低溫氮氣吸附-脫附、H2程序升溫還原(H2-TPR)和二氧化碳吸附-脫附(CO2-TPD)等技術(shù)對催化劑進(jìn)行了表征.結(jié)果顯示:CaO改性對CuZnAlZr催化劑的織構(gòu)性質(zhì)沒有明顯的影響;但共沉淀法制備的CaO改性CuZnAlZr催化劑具有弱堿性和中等強度兩種堿性中心,且有較多的堿中心數(shù)量;而未改性的CuZnAlZr催化劑只具有弱堿性中心.CO加氫反應(yīng)結(jié)果表明:經(jīng)共沉淀法制備的CaO改性CuZnAlZr催化劑對C2+醇的生成有明顯的促進(jìn)作用.
CaO改性;CuZnAlZr催化劑;合成氣;低碳醇
1 引言
隨著人類對油類能源需求的不斷增加以及環(huán)境保護標(biāo)準(zhǔn)的日益嚴(yán)格,尋找高效清潔的燃料替代品和添加劑(可有效提高辛烷值)受到了越來越多的關(guān)注.1-4低碳混合醇是一種C2-C5醇的混合物,它不僅可以作為清潔的燃料添加劑,而且還是許多化學(xué)品和聚合物合成的原料.合成低碳醇的原料通常是合成氣,而合成氣可以通過天然氣重整和煤的氣化獲得,5-13來源十分豐富;而且從長遠(yuǎn)來看,通過生物質(zhì)資源來獲得合成氣,然后將合成氣制成低碳醇或者F-T (Fischer-Tropsch)產(chǎn)品將是一條可持續(xù)發(fā)展的路線.14
合成氣制低碳醇反應(yīng)所需催化劑的組成通常都比較復(fù)雜,3,15-17但是大體上可以分為三類:18(1) F-T合成催化劑組分改性Cu基催化劑;(2)堿性金屬摻雜硫化鉬基催化劑;(3)金屬碳化物催化劑.其中,含F(xiàn)-T合成催化劑活性組分的Cu基催化劑(例如Cu-Co催化劑)被認(rèn)為是有競爭力的一類催化劑.19,20但是這類催化劑尚存在低溫活性差、醇類(尤其是C2+醇)選擇性低、活性組分易燒結(jié)等問題,21-23因此,研制在低溫低壓下具有高活性和高C2+醇選擇性的催化劑是該領(lǐng)域的研究重點之一.
Epling等24研究發(fā)現(xiàn),在CuMgCe氧化物催化劑上進(jìn)行CO加氫反應(yīng)時,在較低的溫度(320°C)和壓力(5 MPa)下,異丁醇的選擇性較高,但其活性比較低.Iglesia等25的研究結(jié)果表明,該催化劑可以看作雙功能催化劑,它有兩種活性中心,即催化CO加氫生成甲醇的Cu活性中心,和催化甲醇進(jìn)行縮合生成更高級醇的MgO堿性活性中心,而CeO2只是起到結(jié)構(gòu)助劑的作用.另一方面,CuZnAl氧化物是一種經(jīng)典的甲醇合成催化劑,26,27而經(jīng)過Zr改性的CuZnAlZr氧化物催化劑有更高的合成甲醇活性和穩(wěn)定性.28-30本實驗室在前期研究中將CuZnAlZr氧化物與堿性氧化物MgO復(fù)合,制備的催化劑對CO加氫制備低碳醇產(chǎn)物中C2+醇的分布有一定的改善作用.31因此,本文進(jìn)一步將CuZnAlZr氧化物與堿土金屬Ca的氧化物進(jìn)行復(fù)合,考察了復(fù)合方式對催化劑性能的影響,以期獲得能夠在較溫和條件下合成出性能優(yōu)異的低碳醇催化劑.
2 實驗部分
2.1 催化劑的制備
所用試劑除特別說明外均為北京化工廠的分析純試劑.
采用共沉淀法和球磨機械混合法制備Ca改性的CuZnAlZr氧化物催化劑.(1)共沉淀法制備CuZnAlZr氧化物催化劑的步驟為:按照Cu:Zn:Al: Zr摩爾比等于0.45:0.35:0.1:0.1稱取相應(yīng)量的金屬硝酸鹽,配制成0.4 mol·L-1的水溶液;用碳酸鉀和氫氧化鉀溶液作為沉淀劑,按摩爾比為2:1稱取適量的無水碳酸鉀和氫氧化鉀,配制成金屬離子濃度為0.375 mol·L-1的水溶液;在攪拌狀態(tài)下將上述兩種溶液并流滴進(jìn)含有一定量沉淀劑的燒杯中進(jìn)行共沉淀,沉淀液溫度保持在60°C,pH保持在9左右;沉淀結(jié)束后,所得的沉淀物經(jīng)2 h老化,去離子水洗滌、離心分離反復(fù)5次,以洗去Na離子;再用乙醇洗滌2次,將水溶膠置換成醇溶膠;最后再次離心,把固體于110°C干燥12 h,于450°C焙燒3 h(升溫速率2°C·min-1),得到CuZnAlZr催化劑.(2)共沉淀法制備CuZnAlZr-CaO復(fù)合催化劑的步驟為:按(CuZnAlZr):CaO的質(zhì)量比為90:10投料(用硝酸鹽配制各硝酸鹽溶液.Ca(NO3)2·4H2O為分析純,北京市紅星化工廠),其它條件和步驟同上述(1),制得的催化劑標(biāo)記為CuZnAlZr-CaO-(CP).(3)機械混合法制備CuZnAlZr-CaO復(fù)合催化劑的步驟為:使用上述(1)中制得的CuZnAlZr復(fù)合氧化物,將一定質(zhì)量的CuZnAlZr和CaO(按CuZnAlZr:CaO的質(zhì)量比為90:10)置于容積為100 mL的球磨罐中,并向其中加入無水乙醇;將球磨罐裝于行星式球磨機(南京大學(xué)儀器廠,QM-3SP04型)中進(jìn)行球磨(公轉(zhuǎn)速率為300 r·min-1,自轉(zhuǎn)速率600 r·min-1,球磨時間6 h),所得漿狀物經(jīng)離心分離后于110°C干燥12 h,于450°C焙燒3 h(升溫速率2°C·min-1),得到機械混合法制備的CuZnAlZr-CaO催化劑,標(biāo)記為CuZnAlZr-CaO-(BM).
2.2 催化劑的表征
催化劑晶相結(jié)構(gòu)的測定使用德國Brüker公司的D8 Advance型X射線衍射儀進(jìn)行測定.以CuKα射線為輻射源,管電壓為40 kV,管電流為40 mA,掃描步進(jìn)0.02°.催化劑織構(gòu)性質(zhì)的測定采用N2物理吸附-脫附法,所用儀器為Micromeritics ASAP-2010C型吸附儀.樣品測試之前先在200°C下脫氣5 h,然后采用低溫(-196°C)N2吸附法測定.樣品的比表面積是根據(jù)N2物理吸附等溫線,采用BET方法計算得到;孔體積以吸附曲線上相對壓力為0.995時的氣體吸附量進(jìn)行計算;平均孔徑及孔徑分布根據(jù)吸附等溫線的脫附支,采用BJH方法計算.催化劑的還原性質(zhì)由H2的程序升溫還原方法進(jìn)行表征.H2程序升溫還原具體步驟為:稱取樣品0.1 g置于U形管中,在高純He氣流(30 mL·min-1)中以15°C·min-1升至400°C并保持1 h進(jìn)行預(yù)處理,以除去樣品表面吸附的水及其它雜質(zhì),然后在He氣流中降至室溫,切換為H2/Ar(V/V=5/95)混合氣(30 mL·min-1),待基線穩(wěn)定后以15°C·min-1的升溫速率開始升溫至600°C,熱導(dǎo)檢測器檢測氫氣隨溫度變化的消耗情況.催化劑表面的堿性質(zhì)由CO2的程序升溫脫附方法進(jìn)行表征,具體實驗步驟為:稱取樣品0.1 g置于U形管中,在高純He氣流(30 mL·min-1)中以15°C· min-1升至750°C并保持1 h進(jìn)行預(yù)處理,以除去樣品表面吸附的水及其它雜質(zhì),然后在He氣流中(30 mL·min-1)降溫至100°C,切換為高純CO2氣流(30 mL·min-1),吸附30 min,然后再切換為He氣流,于100°C下吹掃30 min.待基線穩(wěn)定后,在He氣流中以15°C·min-1的升溫速率由100°C開始程序升溫至800°C;脫附的CO2采用AMETEK公司的DM 200m型四極質(zhì)譜儀進(jìn)行定量檢測.H2-TPR及CO2-TPD測試所用儀器為Quantachrome公司的CHEMBET 3000 TPR/TPD吸附儀和DM 200m型四極質(zhì)譜儀.
2.3 催化劑的性能測試
CO加氫制低碳醇反應(yīng)在微型高壓固定床反應(yīng)裝置(北京欣航盾科技有限公司制造)上進(jìn)行.所采用的反應(yīng)器為不銹鋼管內(nèi)襯石英管,石英管內(nèi)徑10 mm;催化劑填充量為0.5 g(以CuZnAlZr質(zhì)量計),反應(yīng)前,催化劑用50 mL·min-1的H2/Ar混合氣(體積比為10/40)在270°C下還原4 h(升溫速率為3°C· min-1);然后將溫度降至150°C以下,切換為合成氣(H2/CO=1/1),調(diào)整體系壓力為6.0 MPa,調(diào)節(jié)合成氣流速,穩(wěn)定一段時間后再升溫至所需反應(yīng)溫度,空速為6000 mL·h-1·g-1(催化劑量以CuZnAlZr的質(zhì)量計).反應(yīng)產(chǎn)物用兩臺氣相色譜聯(lián)合進(jìn)行在線檢測和分析.TDX-01色譜柱分離N2、CO、CH4和CO2,TCD檢測器定量檢測;Al2O3毛細(xì)管柱(?0.53 mm×30 m)分離烴類產(chǎn)物,氫火焰離子化檢測器(FID)定量檢測TDX-01、PQ和OV-17色譜柱組合分離的烴類(主要是甲烷)和含氧化合物.
3 結(jié)果與討論
3.1 催化劑的織構(gòu)參數(shù)
圖1為CuZnAlZr、CuZnAlZr-CaO-(BM)和CuZnAlZr-CaO-(CP)催化劑的等溫N2吸附-脫附曲線和孔徑分布圖.由圖可以看出,所有樣品的吸附等溫線均為IV類吸附等溫線,說明這些催化劑含有介孔結(jié)構(gòu),有較大的比表面積;另外,各樣品等溫N2吸附-脫附曲線上回滯環(huán)閉合點的位置比較靠近高相對壓力端,由此可以推斷這些催化劑的平均孔徑均較大.從孔徑分布圖上看,各催化劑的孔徑分布沒有明顯變化,只是其孔體積在添加CaO后有所減小,順序為 CuZnAlZr>CuZnAlZr-CaO-(CP)>CuZnAlZr-CaO-(BM).

圖1 催化劑的N2吸附-脫附及孔徑分布曲線Fig.1 Nitrogen adsorption-desorption isotherms and pore size distribution of catalystsBM:mechanicalmixing method;CP:co-precipitation method

表1 催化劑的織構(gòu)參數(shù)Table 1 Texture parameters of catalysts
表1列出了催化劑的織構(gòu)參數(shù).與CuZnAlZr催化劑相比,添加CaO改性后催化劑的比表面積有所降低,由未經(jīng)改性的CuZnAlZr催化劑的138.0 m2· g-1分別降至了機械混合法改性的CuZnAlZr-CaO-(BM)催化劑的78.0 m2·g-1和共沉淀法改性催化劑CuZnAlZr-CaO-(CP)的85.9 m2·g-1;CaO改性后的CuZnAlZr催化劑的孔體積有所減小,尤其是機械混合法制備的CaO改性催化劑CuZnAlZr-CaO-(BM),其孔體積降低了50%以上.
3.2 催化劑的晶相結(jié)構(gòu)
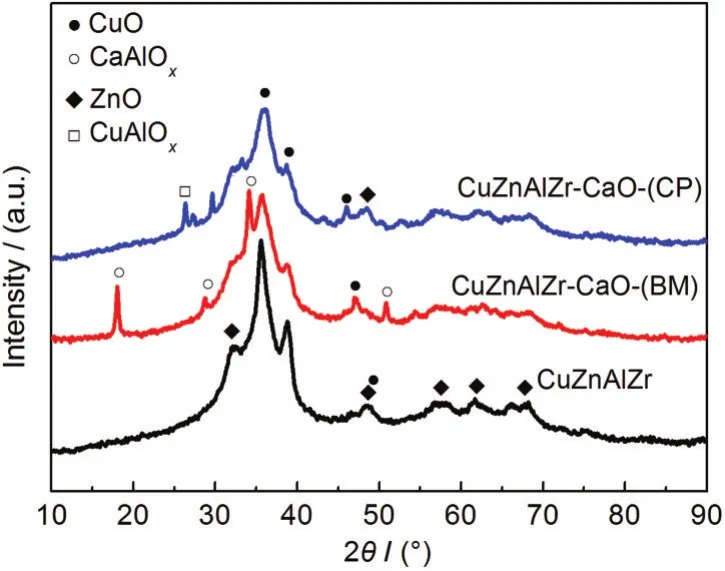
圖2 催化劑的XRD譜圖Fig.2 XRD patterns of catalysts
圖2為CuZnAlZr催化劑和CaO改性CuZnAlZr催化劑的XRD圖譜.由圖可見,未經(jīng)改性的CuZnAlZr催化劑的XRD譜圖上僅出現(xiàn)了CuO(35.6°、38.7°、48.4°)的衍射峰和較為彌散的ZnO衍射峰(32.2°、 48.4°、56.8°、61.4°、68.1°);而由于Al2O3和ZrO2的含量較低,因此未觀察到它們的衍射峰,說明它們以無定形或微晶狀態(tài)存在,分散得較好.在CaO改性后的CuZnAlZr催化劑中,CuO衍射峰有所寬化,衍射強度逐漸減弱(CuZnAlZr>CuZnAlZr-CaO-(CP)>CuZnAlZr-CaO-(BM)),說明CuO晶粒變小,其分散程度得到了改善;在經(jīng)CaO改性后的催化劑中出現(xiàn)了CaAlOx物相,且與機械混合法制備的催化劑相比,在用共沉淀法制備的改性催化劑上,其衍射峰強度相對較弱,說明CaAlOx晶粒較小.從圖中還可以看出,在經(jīng)CaO改性后的CuZnAlZr催化劑中, ZnO的衍射峰都有不同程度的寬化,說明其分散程度也隨著助劑的添加得到了改善.

圖3 催化劑的H2-TPR譜圖Fig.3 H2-TPR profiles of catalysts
3.3 催化劑的H2-TPR表征
圖3是CuZnAlZr催化劑及經(jīng)CaO改性的CuZnAlZr催化劑的H2-TPR圖.由圖可見,三個樣品都存在大而寬的CuO還原峰(CuZnAlZr催化劑在150-450°C,經(jīng)CaO改性的催化劑在180-385°C),但是經(jīng)CaO改性催化劑的還原峰面積有大幅度減小,這一方面是因為CaO的加入減少了單位質(zhì)量樣品中CuO的含量,另一方面可能是因為CaO的加入使得CuO與載體之間的相互作用增強,使得部分CuO難以被還原.從表2中的數(shù)據(jù)可以看出,三種催化劑中CuO的還原程度順序為CuZnAlZr>CuZnAl-Zr-CaO-(CP)>CuZnAlZr-CaO-(BM).
3.4 催化劑的CO2-TPD表征
圖4是三種催化劑樣品的CO2-TPD圖.由圖可以看出CuZnAlZr催化劑僅在172°C左右出現(xiàn)一個CO2脫附峰,而添加了CaO后的CuZnAlZr-CaO催化劑分別在180和496°C左右出現(xiàn)兩個CO2的脫附峰,并且經(jīng)過CaO改性的催化劑的低溫脫附峰的溫度稍高于未改性催化劑的CO2脫附峰溫度;添加了CaO的催化劑在低溫區(qū)的脫附峰面積大于未改性的催化劑,而且用共沉淀法制備的CuZnAlZr-CaO-(CP)催化劑在高溫區(qū)的CO2脫附峰也明顯大于用機械混合法制備的CuZnAlZr-CaO-(BM)催化劑的脫附峰.如果將低溫區(qū)脫附峰歸屬于弱堿性位吸附的CO2,將高溫區(qū)脫附峰歸屬于中強堿性位吸附的CO2,則通過計算可以看出(計算結(jié)果見表3): CuZnAlZr催化劑僅存在少量的弱堿性位;經(jīng)CaO改性后催化劑的弱堿性位數(shù)量有了較大幅度的增加;而且在CaO改性后的催化劑表面出現(xiàn)了中強堿性位,其中CuZnAlZr-CaO-(CP)催化劑上的中強堿性位數(shù)量要多于CuZnAlZr-CaO-(BM)上的中強堿性位.這說明催化劑的堿性位數(shù)量及堿強度順序為CuZnAlZr-CaO-(CP)>CuZnAlZr-CaO-(BM)>CuZnAlZr,即CuZnAlZr-CaO-(CP)催化劑的總堿性位在三個催化劑中是最多的,而且其中強堿性位含量也是最多的.

表2 催化劑的相對還原程度Table 2 Relative reduction degree of catalysts

圖4 催化劑的CO2-TPD譜圖Fig.4 CO2-TPD profiles of catalysts
3.5 催化劑的CO加氫反應(yīng)性能
在本文所采用的反應(yīng)條件下,Cu基催化劑催化CO加氫反應(yīng)的產(chǎn)物包括低碳醇(C1-C5醇)、低碳烴和CO2等.表4和表5分別列出了CuZnAlZr催化劑以及CaO改性的催化劑在CO加氫制低碳醇反應(yīng)中的性能.從表中數(shù)據(jù)可以看出,在反應(yīng)溫度為300°C時,三個催化劑的CO轉(zhuǎn)化率在22.3%-26.5%范圍內(nèi),其中CuZnAlZr-CaO-(CP)的CO轉(zhuǎn)化率稍高些;在CuZnAlZr-CaO-(BM)上的總醇(ROH)選擇性略有提高,并且總醇中的甲醇含量分布比其它兩個催化劑也要高一些,這可能是和CuZnAlZr-CaO-(BM)的堿性較弱,生成的甲醇物種未能充分縮合生成高級醇有關(guān).采用共沉淀法制備的CaO改性催化劑CuZnAlZr-CaO-(CP)上甲醇在總醇中的分布有所降低,這可能是因為其較強的堿性位較多,有利于催化劑上生成的甲醇物種縮合成高級醇,相應(yīng)的C2+醇在總醇中的含量得到了提高(C2+醇選擇性由35.7%提高到了44.7%),C2+醇的時空收率(STY)也由原來的67.6 g·kg-1·h-1提高到了90.7 g·kg-1·h-1.

表3 催化劑的堿性質(zhì)Table 3 Basic properties of catalysts
由表6可知,當(dāng)反應(yīng)溫度為350°C時,三個催化劑的CO轉(zhuǎn)化率處于37.2%-42.6%范圍內(nèi),其中CuZnAlZr-CaO-(BM)催化劑的活性較低;而比表面積稍高的CuZnAlZr-CaO-(CP)催化劑CO轉(zhuǎn)化率稍高些(可達(dá)42.6%).但是烴類和醇類選擇性與反應(yīng)溫度為300°C時的值相比都有所降低.與在300°C下反應(yīng)結(jié)果相似,采用共沉淀法制備的CaO改性催化劑CuZnAlZr-CaO-(CP)上C2+醇在總醇中的含量提高了(其選擇性由55.9%提高到了64.9%),相應(yīng)的C2+醇的時空收率(STY)也達(dá)到了175.3 g·kg-1·h-1,與未改性的CuZnAlZr催化劑相比有了較大幅度的提高.
從表5和表7中可以看出,在CaO改性CuZnAlZr催化劑上低碳醇的產(chǎn)物中,除甲醇外以C3和C4醇類為主,具有典型的改性甲醇合成催化劑的產(chǎn)物分布特點.32在300和350°C時CuZnAlZr-CaO-(BM)催化劑上甲醇選擇性都比較高,而C3-C5醇的選擇性分別都有所降低.這可能是因為在中強堿性位上有利于甲醇物種縮合生成高級醇,因此與CuZnAlZr-CaO-(CP)催化劑相比,CuZnAlZr-CaO-(BM)上C3-C5醇選擇性要低;同時還可能由于CuZnAlZr-CaO-(BM)催化劑活性較低,生成的甲醇物種在催化劑表面濃度也較低,造成甲醇物種縮合的機率降低,導(dǎo)致其對C2+醇選擇性也要低一些.

表4 不同催化劑在300°C時的CO加氫反應(yīng)性能Table 4 Performance of different catalysts in CO hydrogenation reaction at 300°C

表5 不同催化劑在300°C時的醇類產(chǎn)物分布Table 5 Distribution of alcohols over different catalysts at 300°C
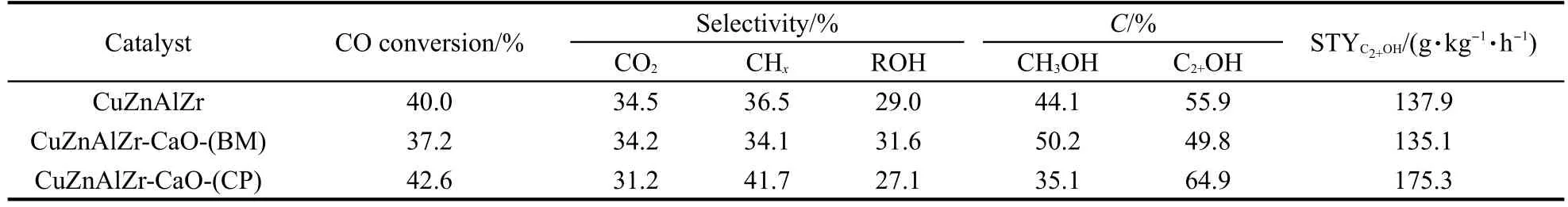
表6 不同催化劑在350°C時的CO加氫反應(yīng)性能Table 6 Performance of different catalysts in CO hydrogenation reaction at 350°C

表7 不同催化劑在350°C時的醇類產(chǎn)物分布Table 7 Distribution of alcohols over different catalysts at 350°C
從以上結(jié)果可以看出,CuZnAlZr催化劑本身對CO加氫生成甲醇的活性較高,反應(yīng)條件也較為溫和,只是其生成C2+醇的能力不足.在添加了助劑CaO后,催化劑中CuO的還原程度有所降低,進(jìn)一步促進(jìn)了甲醇生成的活性,這與Fujitani等33的研究結(jié)果一致,他們認(rèn)為:H2和CO合成甲醇的反應(yīng)是在Cu-CuO界面上進(jìn)行的,純銅對甲醇合成是沒有活性的,因此CuO的過度還原反而不利于甲醇的生成.由于CuZnAlZr-CaO-(BM)催化劑的比表面小,在相同反應(yīng)條件下CO加氫活性較差,堿性弱且堿性位也少,這可能不利于甲醇物種縮合生成C2+醇.因此在CuZnAlZr-CaO-(BM)催化劑上甲醇選擇性較高而C2+醇選擇性較低.用共沉淀法制備的CuZnAlZr-CaO-(CP)催化劑上有較多的堿性位和較強的堿性,這使生成的甲醇物種更容易縮合生成更高級的醇,增加了C2+醇在總醇中的比例,從而使C2+醇的收率得到了提高.
4 結(jié)論
在CO加氫合成低碳醇反應(yīng)中,CaO的添加對CuZnAlZr-CaO催化劑的催化性能有顯著的影響,通過共沉淀法添加CaO制備的CuZnAlZr-CaO-(CP)催化劑上有更多的堿性位,使得生成的甲醇物種更容易縮合生成更高級的醇,提高了C2+醇選擇性和收率,從而可顯著提高CuZnAlZr催化劑C2+醇在總醇中的含量.
(1) Li,D.B.;Yang,C.;Li,W.H.;Sun,Y.H.;Zhong,B.Top.Catal. 2005,32,233.
(2) Subramanian,N.D.;Balaji,G.;Kumar,C.S.S.R.;Spivey,J.J. Catal.Today 2009,147,100.
(3) Surisetty,V.R.;Dalai,A.K.;Kozinski,J.Appl.Catal.A 2011, 393,50.
(4) Fang,Y.Z.;Liu,Y.;Zhang,L.H.Appl.Catal.A 2011,397,183.
(5) Gallego,G.S.;Batiot-Dupeyrat,C.;Barrault,J.;Florez,E.; Mondragón,F.Appl.Catal.A 2008,334,251.
(6) San-José-Alonso,D.;Juan-Juan,J.;Illán-Gómez,M.J.;Román-Martínez,M.C.Appl.Catal.A 2009,371,54.
(7) Jakobsen,J.G.;Jakobsen,M.;Chorkendorff,I.;Sehested,J. Catal.Lett.2010,140,90.
(8) Xu,J.H.;Yeung,C.M.Y.;Ni,J.;Meunier,F.;Acerbi,N.; Fowles,M.;Tsang,S.C.Appl.Catal.A 2008,345,119.
(9) Christian Enger,B.;L?deng,R.;Holmen,A.Appl.Catal.A 2008,346,1.
(10) Silva,C.R.B.;da Concei??o,L.;Ribeiro,N.F.P.;Souza,M. M.V.M.Catal.Commun.2011,12,665.
(11) Maestri,M.;Vlachos,D.G.;Beretta,A.;Groppi,G.;Tronconi, E.J.Catal.2008,259,211.
(12) Mortola,V.B.;Damyanova,S.;Zanchet,D.;Bueno,J.M.C. Appl.Catal.B 2011,107,221.
(13) Baek,S.C.;Bae,J.W.;Cheon,J.Y.;Jun,K.W.;Lee,K.Y. Catal.Lett.2011,141,224.
(14) Ye,T.Q.;Zhang,Z.X.;Xu,Y.;Yan,S.Z.;Zhu,J.F.;Liu,Y.;Li, Q.X.Acta Phys.-Chim.Sin.2011,27,1493.[葉同奇,張朝霞,徐 勇,顏世志,朱九方,劉 勇,李全新.物理化學(xué)學(xué)報, 2011,27,1493.]
(15) Shu,L.;Kaliaguine,S.Appl.Catal.B 1998,16,L303.
(16)Mahdavi,V.;Peyrovi,M.H.;Islami,M.;Yegane,M.J.Appl. Catal.A 2005,281,259.
(17) Yang,C.;Li,J.Q.;Cai,F.P.;Sun,L.;Wu,J.H.Preparation and Application of Catalysts for HigherAlcohols Synthesis from Syngas.CN Patent 101653729,2010-02-24.[楊 成,李建青,蔡飛鵬,孫 立,吳晉滬.一種用于合成氣制低碳醇的催化劑及制法和應(yīng)用:中國,CN101653729[P].2010-02-24.]
(18)Fang,K.G.;Li,D.B.;Lin,M.G.;Xiang,M.L.;Wei,W.;Sun, Y.H.Catal.Today 2009,147,133
(19) Shi,L.M.;Chu,W.;Liu,Z.C.Chem.Ind.Eng.Prog.2011,30, 162.[士麗敏,儲 偉,劉增超.化工進(jìn)展,2011,30,162.]
(20) Gupta,M.;Smith,M.L.;Spivey,J.J.ACS Catal.2011,1,641.
(21) Apesteguia,C.R.;De Rites,B.;Miseo,S.;Soled,S.Catal.Lett. 1997,44,1.
(22)Lin,M.G.;Fang,K.G.;Li,D.B.;Sun,Y.H.Catal.Commun. 2008,9,869.
(23)Yang,X.M.;Wei,Y.;Su,Y.L.;Zhou,L.P.Fuel Process. Technol.2010,91,1168.
(24)Epling,W.S.;Hoflund,G.B.;Hart,W.M.;Minahan,D.M. J.Catal.1997,169,438.
(25) Xu,M.T.;Gines,M.J.;Hilmen,A.M.;Stephens,B.L.;Iglesia, E.J.Catal.1997,171,130.
(26) Jurgen,L.J.;Koy,J.;Regula,T.Catalyst for Methanol Synthesis.US.Pat.Appl.7754651[P].2002-7-13.
(27)Wang,L.L.;Yang,L.M.;Zhang,Y.H.;Ding,W.;Chen,S.P.; Fang,W.P.;Yang,Y.Q.Fuel Process.Technol.2010,91,723.
(28) An,X.;Li,J.L.;Zuo,Y.Z.;Zhang,Q.Catal Lett.2007,118, 264.
(29) Lim,H.W.;Park,M.J.;Kang,S.H.;Chae,H.J.;Bae,J.W.; Jun,K.W.Ind.Eng.Chem.Res.2009,48,10448.
(30) Zhang,Q.;Zuo,Y.Z.;Han,M.H.;Wang,J.F.;Jin,Y.;Wei,F. Catal.Today 2010,150,55.
(31) Zhang,R.J.Research on the Production of Isobutene and Isobutanol from CO Hydrogenation.Ph.D.Dissertation, Tsinghua University,Beijing,2011.[張榮俊.CO加氫異構(gòu)合成制異丁烯和異丁醇的研究[D].北京:清華大學(xué),2011.]
(32) Xu,M.T.;Iglesia,E.Catal.Lett.1998,51,47.
(33) Fujitani,T.;Nakamura,J.Catal.Lett.1998,56,119.
January 7,2012;Revised:March 14,2012;Published on Web:March 30,2012.
Effect of CaO Modification on Performance of CuZnAlZr Catalyst in Synthesis of Higher Alcohols from Synthesis Gas
ZHU Qiu-Feng ZHANG Rong-Jun HE De-Hua*
(Innovative Catalysis Program,Key Laboratory of Organic Optoelectronics and Molecular Engineering of Ministry of Education, Department of Chemistry,Tsinghua University,Beijing 100084,P.R.China)
CaO modified CuZnAlZr mixed oxide catalysts were prepared by mechanical mixing and co-precipitation methods,and their catalytic performances for CO hydrogenation to higher alcohols were investigated using a continuousflow high-pressure fixed-bed micro-reactor.The catalystswere characterized by X-ray diffraction(XRD),N2-adsorption-desorpotion,temperature-programmed reduction of H2(H2-TPR),and temperature-programmed desorption of CO2(CO2-TPD).The results showed that doping with CaO did not alter the textural properties of the catalyst,but increased the number of basic sites. Furthermore,the CaO-modified CuZnAlZr catalysts possessed both weak and medium strength basic sites,while unmodified CuZnAlZr catalyst possessed only weak basic sites.Catalytic performance, assessed by CO hydrogenation to C2+alcohols,was improved in the CaO-doped CuZnAlZr catalyst relative to the undoped catalyst.
CaO modification;CuZnAlZr catalyst;Synthesis gas;Higher alcohol
10.3866/PKU.WHXB201203302
?Corresponding author.Email:hedeh@mail.tsinghua.edu.cn;Tel:+86-10-62773346.
The project was supported by the National High Technology Research and Development Program of China(863)(2007AA05Z332).
國家高技術(shù)研究發(fā)展計劃(863)(2007AA05Z332)資助項目
O643
猜你喜歡
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
中國塑料(2016年12期)2016-06-15 20:30:07
浙江大學(xué)學(xué)報(工學(xué)版)(2016年11期)2016-06-05 09:21:04
中國塑料(2016年5期)2016-04-16 05:25:36
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
