五條蚋(Simulium quinquestriatum)不同地理種群遺傳多樣性的ISSR標記研究
2013-09-13 11:46:58修江帆張春林陳漢彬
生物技術通報 2013年7期
關鍵詞:物種
修江帆 張春林 陳漢彬
五條蚋 Simulium(Simulium)quinquestriatum Shiraki,1935,隸屬于蚋科(Simuliidae)蚋屬(Simulium)蚋亞屬(Simulium)盾紋組(stiatum-group),廣泛分布于東洋界,現(xiàn)已報道的國家有中國、韓國、日本、泰國和越南等[1]。在我國幾乎全境皆有分布,已見記錄的省區(qū)有臺灣、遼寧、福建、江西、陜西、廣東、廣西、湖南、貴州、四川、云南、西藏等[2]。目前,五條蚋除吸血、騷擾人畜外,其流行病學尚不清楚,在此方面研究報道非常少見,僅見Fukudam于2008年報道五條蚋幼蟲能夠試驗感染微絲蚴(microfilariae)[3]。據(jù)觀察,五條蚋一般分布于人畜活動頻繁的地區(qū),對生境的適應范圍廣,偏好滋生于有機質(zhì)含量豐富的水體,往往是該地區(qū)的優(yōu)勢蚋種。因此認為五條蚋的分布范圍廣,適應性強,易于采集,是研究蚋類種群演化及遺傳多樣性的良好材料。
簡單重復序列間區(qū)(Inter-Simple Sequence Repeat,ISSR)標記技術是由加拿大蒙特利爾大學Zietkiewicz等[4]于1994年在微衛(wèi)星標記的基礎上發(fā)展起來的一種分子標記技術。該標記技術綜合了其它標記技術的多種優(yōu)點,具有操作簡單、引物開發(fā)費用低、穩(wěn)定性好、檢測多態(tài)性能力強、所需DNA模板量少、無需知道基因組序列等特點,已被成功地運用于親緣關系、遺傳多樣性、種質(zhì)鑒定和構(gòu)建遺傳圖譜等研究領域[5-9]。關于蚋類的ISSR相關研究僅見于捷克斯洛伐克Dusinsky[10]關于蚋種內(nèi)和種間的鑒定,及本課題組廉國盛[11]關于中國8亞屬23蚋種ISSR分子進化研究;而蚋類種群ISSR研究在國內(nèi)外均未見相關報道。本研究采用ISSR分子標記技術對中國8個五條蚋種地理群種的遺傳多樣性及遺傳分化進行研究分析,旨在從分子水平對五條蚋種群遺傳結(jié)構(gòu)及演化的研究提供基礎資料。
1 材料與方法
1.1 材料
試驗用五條蚋采自貴州貴陽三江(GZSJ)、貴州青巖(GZQY)、貴州息烽(GZXF)、福建長樂(FJCL)、福建寧德(FJND)、四川夾江(SCJJ)、廣西興安(GXXA)、云南勐臘(YNML)5省8個地理種群(表1),每個種群分別取20條個體進行基因組DNA提取,-20℃凍存?zhèn)溆谩?/p>
試驗主要藥品:引物由上海生工生物工程有限公司合成;dNTP,Mg2+,Taq DNA聚合酶,Marker DL2000 DNA Ladder 購置于TaKaRa公司;BSA(牛血清白蛋白)購置于Sigma公司。

表1 用于ISSR分析的五條蚋種群
1.2 方法
1.2.1 基因組DNA的提取及定量保存 參照酚-氯仿抽提法[12],每個種群選取20條五條蚋提取基因組DNA,8個種群共計160個DNA樣本,經(jīng)電泳檢測后,用紫外分光光度計將其濃度定量為50ng/μL,-20℃保存。
1.2.2 ISSR分析
1.2.2.1 引物的篩選 經(jīng)前期試驗優(yōu)化設計的五條蚋ISSR-PCR最佳反應體系中篩選出多態(tài)性高、穩(wěn)定性好、條帶清晰的8條引物用于PCR擴增反應(表 2)。
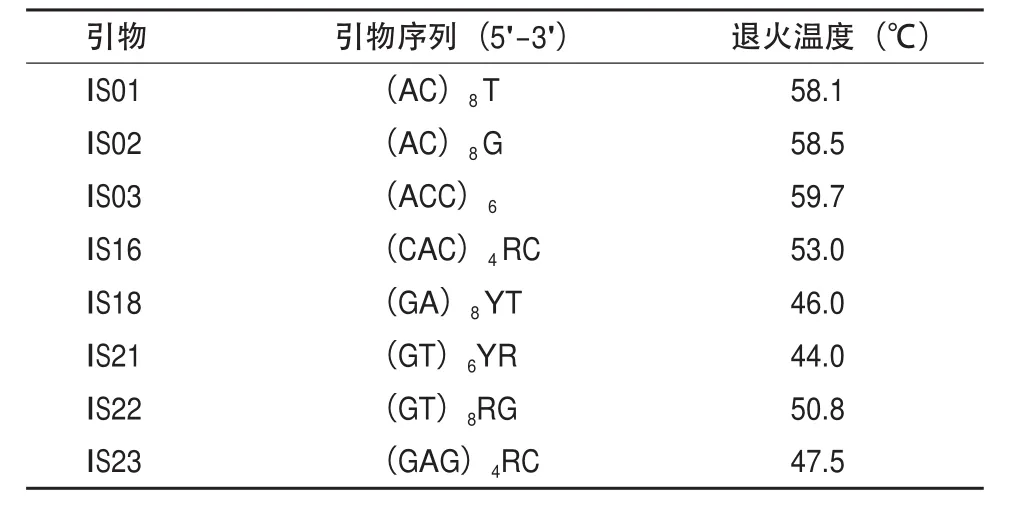
表2 ISSR-PCR 擴增所用引物
1.2.2.2 PCR 擴增 20μL 反應體系中引物1.0μmol/L、模板 50.0ng/μL、dNTP0.15mmol/L、Mg2+1.50mmol/L、BSA 2.00mg/mL、Taq DNA 聚合酶 0.15 5U/μL,ddH2O補齊。PCR反應在5331(德國Eppendorf)基因擴增儀中進行,PCR反應程序為:94℃預變性5min;94℃變性 50s,51℃退火 60s,72℃延伸 90s,35個循環(huán);72℃延長10min,10℃保存[11]。PCR產(chǎn)物與1μL 6×loading buffer混勻后取 6μL,以 TaKaRa公司的DL2000 Marker為對照,用1.0%瓊脂糖凝膠(含EB0.05μg/mL),電泳緩沖液為0.5×TBE,電壓200 V恒定電泳40min,凝膠成像系統(tǒng)拍照記錄。
1.2.3 數(shù)據(jù)統(tǒng)計與分析 運用Quantity One 4.2.1軟件分析ISSR擴增的譜帶,在相同遷移位置上,有譜帶存在賦值為“1”,無譜帶賦值為“0”,建立五條蚋8個種群的二元數(shù)據(jù)矩陣。將數(shù)據(jù)導入POPGEN1.32軟件[13],計算五條蚋不同種群的遺傳距離,Nei氏基因多樣性指數(shù)H(gene diverdity),Shannon 氏信息指數(shù) I(shannon’s information index),多態(tài)性位點比例P(percentage of polymorphic loci),同位點等位基因數(shù)A(Observedmean number of alleles per locus),同位點有效等位基因數(shù)AE(Effectivemean number of alleles per locus),遺傳分化系數(shù)(Coefficient of gene differentiation,Gst)[14]。 運 用 軟 件MEGA version 4.0[15]選擇非加權(quán)組平均法UPGMA(unweighted pair-groupmethod for arithmetic averages analysis)和鄰位聚類法NJ(Neighbor-Joining)對五條蚋8個種群進行種群聚類分析。Google earth 6.0.1軟件計算8個種群的地理距離。GenAlEx 6.41軟件對種群間和種群內(nèi)的遺傳變異進行分子方差分析AMOVA[16](Analysis of Molecular Variance),ISSR 表型特征的主成分分析(PCA主成分分析)[17]并檢測種群間地理距離與遺傳距離的相關性。
2 結(jié)果
2.1 五條蚋種群DNA的ISSR擴增結(jié)果
篩選的8條引物對8個種群共160條五條蚋DNA進行ISSR-PCR擴增,共獲得111條有效條帶,這些條帶的大小介于200-2000bp之間(圖1),其中引物IS18擴增條帶數(shù)最多,為20條;引物IS22擴增條帶數(shù)最少,為9條,平均每條引物擴增13.88條條帶;不同引物擴增出的多態(tài)性條帶數(shù)和多態(tài)性條帶比例有一定差異,引物IS01多態(tài)性比例最大,為93.33%;引物IS03最小,為76.47%(表3)。不同種群的每一個個體均呈現(xiàn)出為宜的ISSR基因型,這也證明ISSR標記對五條蚋具有很高的鑒別能力,同時也說明五條蚋個體之間存在較大的遺傳變異。這些位點在種群中分布不均勻。從五條蚋8個種群的多態(tài)性位點比例可看出五條蚋的遺傳多樣性較為豐富。

圖1 引物IS01對五條蚋群體的ISSR擴增電泳圖

表3 五條蚋種群ISSR擴增引物產(chǎn)生的多態(tài)性條帶
2.2 五條蚋種群的遺傳多樣性
從表4中可看出,五條蚋在物種水平上多態(tài)性位點比例為86.47%,單個種群多態(tài)性條帶比例的變化范圍從最低的種群GZQY(P=66.25%)到最高的種群GXXA(P=86.57%)。在物種水平上同位點有效等位基因AE平均數(shù)為1.553 2±0.334 5,范圍從最低的種群YNML(1.2090±0.2090)到最高的種群GZSJ(1.3811±0.398 6)。Shannon氏多樣性指數(shù)(I)物種水平為0.484 7±0.203 7變化范圍從最低的種群YNML(0.194 3±0.2591)到最高的種群GZSJ(0.314 4±0.305 5)。Nei氏遺傳距離(H)物種水平為0.3231±0.157 9,變化范圍從最低的種群YNML(0.126 4±0.178 4)到最高的種群 GZSJ(0.215 3±0.2141)。總體結(jié)果分析五條蚋物種水平的遺傳多樣性水平比單個種群的高;種群間相比較同位點有效等位基因AE數(shù)、Shannon氏多樣性指數(shù)(I)、Nei氏遺傳距離(H)所反映的變化趨勢一致(YNML<FJCL<GXXA<GZQY<GZXF<FJND<SCJJ<GZSJ),但在多態(tài)性位點比例上卻有所不同(GZQY<FJCL<GZXF<SCJJ<GZSJ<FJND<YNML<GXXA)。

表4 五條蚋種群間的遺傳多樣性指數(shù)
2.3 五條蚋種群的遺傳分化
用POPGENE 1.32計算出的遺傳變異分析結(jié)果(表5)表明,五條蚋種群間存在著一定的遺傳分化。8個種群總的遺傳多樣性Ht=0.3231,其中種群內(nèi)部的遺傳多樣性Hs=0.1801,種群間的基因多樣度(Dst=Ht-Hs)為0.1430,Nei的遺傳分化系數(shù)Gst=0.5900,提示有59%的遺傳分化存在于種群間,41%的遺傳分化存在于種群內(nèi)部,種群間的遺傳分化大于種群內(nèi)部的分化,種群內(nèi)部遺傳分化水平相對較低。種群間基因流[(Nm=0.5(1-Gst)/Gst)]為0.347 5,基因流較小。用AMOVA進行的基于Nei氏距離平方的遺傳變異方差分析結(jié)果也顯示五條蚋的遺傳分化主要存在于種群間,占總分化的59%,種群間的遺傳分化占41%(P<0.001)(表6)。

表5 五條蚋種群遺傳多樣性Nei氏分析

表6 五條蚋種群遺傳分化分子方差分析(AMOVA)
2.4 遺傳距離、地理距離及聚類分析
遺傳距離可以較為直觀地反映每個種群間彼此親緣關系的遠近,為確定五條蚋8個地理種群之間的遺傳關系,對Nei氏遺傳距離進行了計算(表7);根據(jù)采集點的地理坐標計算種群間的地理距離(表7)。五條蚋8個種群中種群GZSJ與種群GZQY之間遺傳距離最近為0.067 6,種群SCJJ和種群YNML之間遺傳距離最遠為0.3410;種群GZSJ與種群GZQY之間地理距離最近為42.31 km,種群FJND和種群YNML之間遺傳距離最遠為908.21 km。
基于種群間Nei氏遺傳距離,運用NJ法和UPGMA法對五條蚋8個種群進行聚類分析。結(jié)果(圖2,圖3)顯示,五條蚋8個種群分化成為2個大組4個亞組,其中種群GZSJ、GZQY、GZXF、SCJJ組成了第1大組,種群FJCL、FJND、GXXA、YNML組成了第2大組。在第1組中,種群GZSJ、GZQY首先聚集形成1支,后又與種群GZXF聚集形成第1亞組,種群SCJJ單獨形成第2亞組;在第2大組中,種群FJCL、FJND匯聚組成第3亞組,種群GXXA、YNML聚集行成第4亞組。NJ法和UPGMA法聚類分析結(jié)果基本一致。PCA主成分分析結(jié)果顯示(圖4),PCA軸1將第一組的4個種群(種群GZSJ、GZQY、GZXF、SCJJ)與第二組的4個種群(種群FJCL、FJND、GXXA、YNML)進行分隔,PCA軸2將第一組中種群GZSJ和種群GZQY聚成一分支,將第二組中的種群FJCL和種群FJND聚成另一分支,種群GZXF單獨聚為一支;PCA的軸3將第一組中種群SCJJ單獨分隔出來成為一亞組,同時將第二組中種群GXXA與種群YNML進行分隔。PCA分析結(jié)果支持NJ法及UPGMA法聚類結(jié)果。

圖2 基于Nei氏遺傳距離做出的五條蚋種群的NJ聚類分析圖

圖3 基于Nei氏遺傳距離做出的五條蚋種群的UPGMA聚類分析圖

圖4 基于ISSR表型特征的主成分分析(PCA)

圖5 五條蚋種群遺傳距離與地理距離的回歸分析
將五條蚋8個種群間的遺傳距離以及地理距離進行相關性的檢驗(mantel test)(圖5),建立回歸方程為y = 7E-05x +0.164 2,相關性系數(shù)| r | =0.510 7(P=0.01<0.05)。結(jié)果說明五條蚋各種群間的遺傳距離與地理距離間呈現(xiàn)正相關,種群間的基因交流可能被它們之間的地理距離所限制。
3 討論
3.1 五條蚋的遺傳多樣性
多態(tài)位點比例(P),Shannon氏多樣性指數(shù)(I),Nei氏基因多樣性指數(shù)(H),同位點上等位基因數(shù)(A)及同位點上有效等位基因數(shù)(AE)是衡量種群多樣性的常用指標[18]。遺傳多樣性是物種或居群長期進化的產(chǎn)物,也是其生存發(fā)展和進化的基礎[19]。一個物種或種群遺傳多樣性水平越高則說明其對環(huán)境變化的適應能力越強。研究對五條蚋8個種群的結(jié)果顯示,五條蚋物種遺傳多樣性水平(AE=1.553 2±0.334 5,H=0.3231±0.157 9,I=0.484 7±0.203 7)較其它昆蟲高,如角倍蚜(AE=1.403±0.338,H=0.247±0.169,I=0.387±0.221)[20];桃蛀螟(AE=1.249 7±0.238 4,H=0.1750±0.1331,I=0.296 6±0.185 5)[21],可說明五條蚋豐富的遺傳多樣性證實其對環(huán)境變化具有較強適應抵抗力的遺傳基礎,這也解釋了五條蚋廣泛分布于東洋界,在我國幾乎全境皆有分布的原因;同時研究發(fā)現(xiàn)五條蚋物種遺傳多樣性水平比單個種群的高,是因其分布廣泛,滋生生境復雜,決定其豐富的遺傳多樣性。
3.2 五條蚋的遺傳分化
生物種群遺傳分化主要決定于種群內(nèi)遺傳漂變與種群間基因流兩者的動態(tài)平衡。Wright 等[22]提出的遺傳分化理論認為:當遺傳分化系數(shù)(Gst)的值介于0-0.05之間表示種群間遺傳分化程度低;0.05-0.15為分化程度中等;0.15-0.25為分化程度高;大于0.25表明種群遺傳分化程度極大。本研究結(jié)果顯示,五條蚋8個種群間的Gst=0.5900,種群分化程度極大;依據(jù)分子方差(AMOVA)分析結(jié)果,五條蚋種群間分化占遺傳分化59%,種群內(nèi)部分化占遺傳分化41%,說明五條蚋的遺傳分化發(fā)生在種群之間,而種群內(nèi)部遺傳分化相對較低,處于次要地位。究其原因:(1)由于地理隔離,導致種群中的遺傳漂變,增加了種群的遺傳多樣性。本研究的五條蚋種群分別采自貴州、云南、四川、廣西、福建5省的8個不同采集點,采集點之間最近相距42.31 km,最遠相距1 908.21 km。地理位置經(jīng)度跨越約18(E:101 34'- E:119 31');緯度跨越約8(N:2128'-N:29 44');海拔 高度差距約1100m(19-1120m);地形有平原、丘陵、盆地和高原。由于采集點之間的地理距離跨度大、地形地貌多樣,可能由于天然地理屏障存在,導致五條蚋的種群間的遺傳分化水平較高。(2)基因流的影響。通常物種基因流的水平高,物種遺傳分化小;基因流的水平低,則物種遺傳分化大。依據(jù)群體遺傳學理論[23],當基因流Nm<1時,就不足以抵制居群內(nèi)因遺傳漂變而引起的居群間遺傳分化;當基因流Nm>4時,表明種群間的基因交流比較充分,足以抵制遺傳漂變的作用,消弱了種群間遺傳分化的產(chǎn)生。五條蚋的Nm=0.347 5<1,說明種群間的基因交流不充分,遺傳漂變的作用較強,可能導致種群間遺傳分化的產(chǎn)生。同時,本研究對種群間的遺傳距離與地理距離的相關性分析,表明8個種群間的遺傳距離在0.126 4-0.215 3之間,距離較大,且種群間的遺傳距離與地理距離呈正相關,地理距離間隔越遠,遺傳距離差距越大。也進一步說明五條蚋種群由于受到地理屏障的制約,不能進行充分的基因交流,種群間基因流水平不高。
3.3 五條蚋種群的聚類分析
本研究基于Nei氏遺傳距離,分別運用NJ法、UPGMA法和PCA主成分分析法對五條蚋8個種群進行聚類分析。3種聚類分析法顯示的結(jié)果均保持一致,表明了聚類分析的結(jié)果可靠性較高。聚類分析的結(jié)果表明,五條蚋8個種群的遺傳距離與它們的地理間隔距離具有一致性。貴州與四川的種群聚成一支,而福建、廣西、云南的種群聚成一支,分別表明貴州與四川的種群之間遺傳距離較近;福建、廣西、云南的種群之間的遺傳距離較近,基因流水平較高,可能代表五條蚋在這些地區(qū)的擴散模式。目前認為蚋類的擴散主要有兩種方式:成蟲的遷飛和幼蟲隨流遷移。據(jù)相關研究報道[24],蚋成蟲的遷飛距離變化較大,如Simulium neavei最大遷飛距離<4 km,而Simulium damnosum及Simulium sirbanum最大遷飛距離可達到400 km。成蟲的遷飛可能是蚋類擴散主要方式,與風向、風力有關,而幼蟲隨流遷移則與水系分布,流速等有關。
4 結(jié)論
構(gòu)建了中國五條蚋8個種群160個體的ISSR指紋圖譜,每個種群均顯示出獨特的圖譜特征。
五條蚋在物種水平遺傳多樣性較高;在種群水平遺傳多樣性較低,五條蚋豐富的遺傳多樣性證實其對環(huán)境變化具有較強適應能力。
五條蚋遺傳分化主要發(fā)生在種群之間,地理屏障(高山和平原等)以及棲息地片段化是導致其遺傳分化形成的主要因素。
[1] Adler PH, Crosskey RW.World Blackflies(Diptera:Simuliidae):A Comprehensive Revision of the Taxonomic and Geographical Inventory [2010].http://entweb.clemson.edu/biomia/pdfs/blackflyinventory.pdf
[2] 陳漢彬, 安繼堯.中國黑蠅(雙翅目:蚋科)[M].北京:科學出版社, 2003:258-259.
[3] Fukuda M, Takaoka H, Uni S, et al.Infective Larve of five Onchocerca species from experimentally infected Simulium species in an area of Zoontic Onchocerciasis in Janpan [J].Parasite, 2008, 15(2):111-119.
[4] Zietkiewicz E, Rafslski A, Labuda D.Genome fingerprinting by simple sequence repeat(SSR)anchored polymerasechain re-action amplification [J].Genomics, 1994, 20:176-183.
[5] 周延清.DNA分子標記技術在植物研究中的應用[M].北京:化工工業(yè)出版社, 2005:68-70.
[6] Agostini G, Echeverrigaray S, Souza-Chies TT.Geneticrela-tionships among South American species of Cunila D.Royen ex L.based on ISSR [J].Plant Syst Evol, 2008, 274:135-141.
[7] Dje Y, Tahi CG, Bi AI, et al.Use of ISSRmarkers to assess genetic diversityof African edibleseeded Citrullus lanatus landraces [J].Scientia Horticulturae, 2010, 124(2):159-164.
[8] Du XY, Zhang QL, Luo ZR.Development of retrotransposon primers and their utilization for germplasm identification in Diospyros spp.(Ebenaceae)[J].Tree Genetics & Genomes, 2009, 5:235-245.
[9] Gupta S, Pandey-Rai S, Srivastava S, et al.Construction of genetic linkagemap of themedicinal and ornamental plant Catharanthus roseus [J].Journal of Genetics, 2007, 86(3):259-268.
[10] Dusinsky R, Kudela M, Stloukalova V, et al.Use of inter-simple sequence respeat(ISSR)markers for discrimination between and within species of blackflies(Diptera:Simuliidae)[J].Section Cellular and Molecular Biology Biologia, 2006, 61(3):299-304.
[11] 廉國盛.山西蚋類區(qū)系研究和中國8亞屬23蚋種ISSR分子進化研究[D].貴陽:貴陽醫(yī)學院,2009.
[12] 范海榮, 夏永靜.四種全血基因組DNA提取方法的比較[J].中國動脈硬化雜志, 2002, 10(6):535-536.
[13] Yeh FC, Yang RC, Boyle T, et al.POPGENE, the user friendly shareware for population genetic analysis [M].Edmonton:University of Alberta, 1997.
[14] Nei M, Li WH.Mathematicalmodel for studying genetic variation in terms of restriction endonucleases [J].Proc Natl Acad Sci USA,1979, 76(10):5269-5273.
[15] Kumar S, Tamura K, Jakobsen IB, et al.MEGA2:Molecular evoutionary genetics analysis software [J].Bioinformatics, 2001,17(12):1244-1245.
[16] 陳俊秋, 慈秀芹.樟科瀕危植物思茅木姜子遺傳多樣性的ISSR分析[J].生物多樣性, 2006, 14(5):410-420.
[17] 王曉明, 賴燕玲.深圳梅林仙湖蘇鐵野生種群遺傳多樣性ISSR分析[J].中山大學學報:自然科學版, 2006, 45(3):82-85.
[18] 朱勛, 楊家強.小菜蛾不同地理種群遺傳多樣性的ISSR標記研究[J].昆蟲學報, 2012, 55(8):981-987.
[19] 陳靈芝.中國的生物多樣性—現(xiàn)狀及其保護對策[M].北京:科學出版社, 1993:11-15.
[20] 王定江, 楊漢遠, 鐘揚, 任竹梅.貴州省八個種群角倍蚜ISSR遺傳多樣性[J].生態(tài)學雜志, 2008, 27(10):1729-1733.
[21] 張穎, 李菁, 王振營.中國桃蛀螟不同地理種群的遺傳多樣性,昆蟲學報, 2010, 53(9):1022-1029.
[22] Wright S.Evolution and the genetics of populations.//Variability within and among natural populations [M].Chicago:University of Chicago Press, 1978.
[23] Slatkin M.Gene flow in natural populations [J].Annual Review of Ecology & Systematics, 1985(16):393-430.
[24] Peter H, Adler A.Evolution, epidemiology, and population genetics of black flies(Diptera:Simuliidae)[J].Genetics and Evolution, 2010(10):846-865.
猜你喜歡
課堂內(nèi)外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內(nèi)外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
課堂內(nèi)外·初中版(科學少年)(2024年12期)2024-12-02 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
中學生博覽(2022年7期)2022-06-21 21:48:14
大科技·百科新說(2021年8期)2021-11-03 10:55:16
學苑創(chuàng)造·A版(2021年5期)2021-06-28 19:51:42
少兒美術(快樂歷史地理)(2020年9期)2020-03-19 05:10:56
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
汽車觀察(2018年10期)2018-11-06 07:05:26
