2,3,5,6-四氟苯基與三氟甲基苯基對噁二唑化合物光電性質影響的理論研究
2014-02-28 06:44:50朱萬強
遵義師范學院學報 2014年2期
關鍵詞:模型
朱萬強,楊 媛
(1.遵義師范學院化學化工學院,貴州遵義563002;2.黔北特色資源應用研究實驗室,貴州遵義563002;3.貴州師范大學化學學院,貴州貴陽550000)
2,3,5,6-四氟苯基與三氟甲基苯基對噁二唑化合物光電性質影響的理論研究
朱萬強1,2,楊 媛3
(1.遵義師范學院化學化工學院,貴州遵義563002;2.黔北特色資源應用研究實驗室,貴州遵義563002;3.貴州師范大學化學學院,貴州貴陽550000)
運用密度泛涵理論B3LYP/6-31G(d)方法,優化了苯環上含氟和三氟甲基的1,3,4-噁二唑類化合物基態的幾何構型,計算了它們相應的紫外吸收光譜。計算結果表明,紫外吸收光譜及分子軌道的能隙值與實驗數據基本吻合;結果還顯示,與不含氟的噁二唑化合物相比,三氟甲基和2,3,5,6-四氟苯單元都能降低化合物的LUMO與HOMO能級,但三氟甲基的影響明顯大于2,3,5,6-四氟苯單基,并且三氟甲基使分子的能隙值增大,分子的吸收和發射光譜發生藍移;而2,3,5,6-四氟苯基卻使分子的能隙值降低,分子的吸收和發射光譜發生紅移,得到與實驗一致的結論。
噁二唑衍生物;2,3,5,6-四氟苯基;三氟甲基苯基;紅外吸收光譜;紫外吸收光譜
近年來,隨著新一代平板顯示器——有機發光二極管的普及和應用,人們更加注重發光材料的高效、節能和長壽命,而這些性能又取決于有機分子的組成、結構和由此決定的發光效率和電子傳輸能力。為此,對有機發光材料進行結構上的修飾,是常采用的方法。對發光材料進行修飾的結果,如能從理論上進行分析和總結,將對今后的研究具有十分積極的指導意義,在這方面的研究已有報道[1-4]。由于1,3,4-噁二唑衍生物具有優良的電子傳輸性、較好的發光性能、熱穩定性及化學穩定性,在電子傳輸材料的應用及研究中,受到廣范的關注[5-10]。對于噁二唑及其衍生物光電性能的理論研究報道不多[11-12]。芳基取代的噁二唑衍生物因其缺電子性、高的熱穩定性和化學穩定性等,已在有機發光材料中得到應用13-15]。為了改善芳基取代的噁二唑衍生物的光電性能,人們已做了多種嘗試[16-19]。用全氟取代稠環芳烴和全氟多連苯來改善有機發光材料的研究,也在近年來受到人們的重視[20-25]。我們曾合成和先后報道了一類新型含氟 2,3,5,6-四氟苯基-以下稱四氟苯基)和三氟甲基芳基取代噁二唑衍生物的光電性質[26-27],但對這類含氟化合物的光電性質理論研究的報道很少看到。在我們先前的報道中,四氟苯基的影響并沒有預期的顯著,僅含四氟苯基分子的HOMO與LUMO能隙值反而降低,導致電化學和光學帶寬紅移,對這一結果,當初沒有得到更好的說明。在本文中,我們對含有三氟甲基和四氟苯基的吸電子取代基噁二唑類衍生物的光電性質從理論上進行了研究,證明了為何四氟苯基沒有如預期地影響雙噁二唑的光電性質,回答了當初的迷惑,希望能為進一步研究設計噁二唑類發光材料和電子傳輸材料提供理論參考。
1 計算模型和方法
采用密度泛函理論(DFT)在B3LYP/6-31G(d)水平上,優化所有分子構型,自洽場收斂標準為程序內定值。振動分析表明所有的優化構型均沒有虛頻,為勢能面上的極小點。并且使用含時密度泛函理論(TD-DFT)計算所有分子構型的電子吸收光譜。全部計算用Gaussian03程序完成[28]。
計算模型如圖1所示。在實驗的基礎上,模型分子M1-M4的光電性質,我們已做過報道[26],其它幾個模型分子的吸收光譜稍后也做了報道[27]。
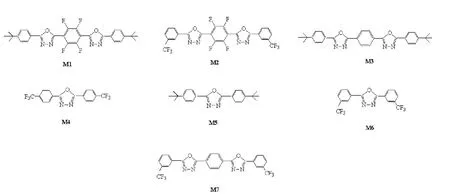
圖1 計算的分子模型
2 結果和討論
2.1 優化構型
通過運用B3LYP/6-31G(d)方法計算,我們得到了模型分子的振動頻率,模型分子的簡振頻率沒有虛頻,說明此構型都是能量極小的值點,是穩定構型。圖2為模型分子的穩定構型。
2.2 模型分子的電子吸收光譜
用計算得到的模型分子的吸收光譜、振子強度以及主要軌道躍遷數據列于表1中,同時與實驗值(均為在CH2Cl2溶劑中測定)作了比較。

圖2 模型分子M1-M7的優化結構
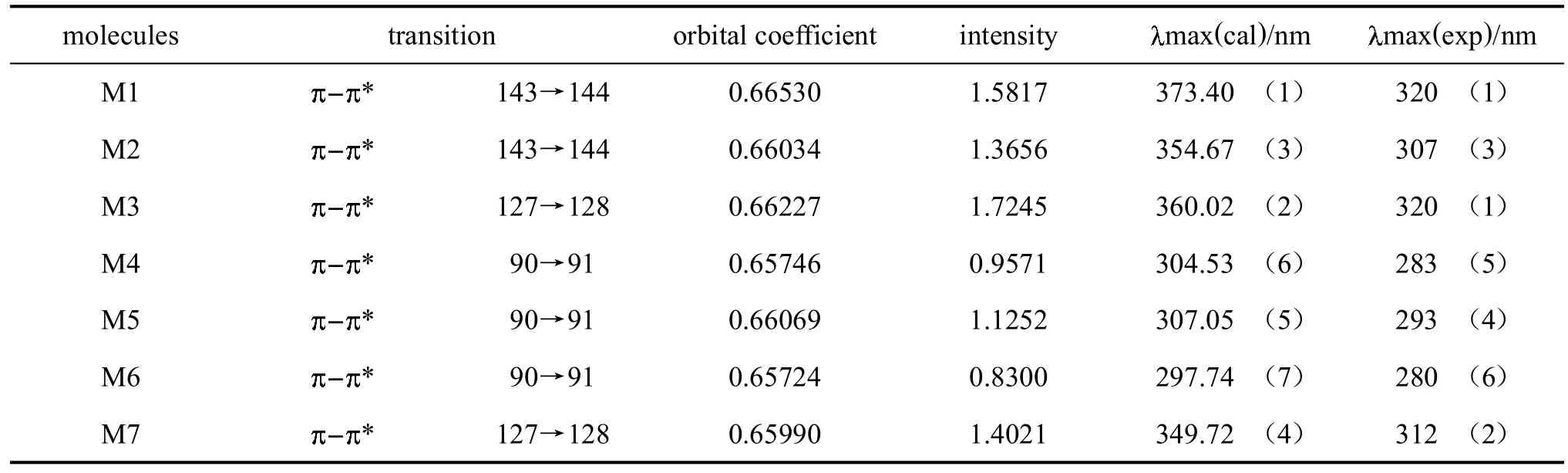
表1 模型分子的吸收光譜
在表1中,計算結果與實驗值分別相差約53、47、40、21、14、17和37nm。雖然有幾個分子的吸收與實驗值相比有較大差距,但考慮到計算值是基于氣相狀態,實驗測定是在CH2Cl2溶液中進行,溶劑效應對結果有一定影響,這樣的誤差還是可接受[29-30],計算的結果是可信的,比如表中數據顯示的吸收光譜的變化趨勢(括號中數字)總體一致。此外M4、M5、M6分子的計算光譜,與實驗值相差值小于或等于21nm,非常吻合。表中數據還顯示,由基態到最低激發單重態的躍遷主要由分子中軌道的垂直躍遷,其它的躍遷強度(未列出)都很小,大部分不到LUMU躍遷的十分之一。吸收光譜的主要躍遷強度,同類型分子M1和M2,M3和M7比較,含三氟甲基的分子(M2,M7),躍遷強度小;不含三氟甲基的分子M1,躍遷強度大;既不含三氟甲基又不含四氟苯基的分子M3,躍遷強度最大;既含三氟甲基又含四氟苯基的分子M2,躍遷強度最小;僅含四氟苯基的分子M1,躍遷強度處于次大位置,從中反映出三氟甲基的影響是主要因素,四氟苯基對分子的電子光譜吸收強度影響不大。在M5與M4、M6三個分子中,含有三氟甲基的躍遷強度也要小得多。
2.3 模型分子的前線軌道分析
2.3.1 軌道能級與分子總能量
在表2中,列出了經計算得到的各個模型分子的前線軌道能量。為了比較同一分子不同構型的能量,還將M2和M7的Trans-構型數據列出。
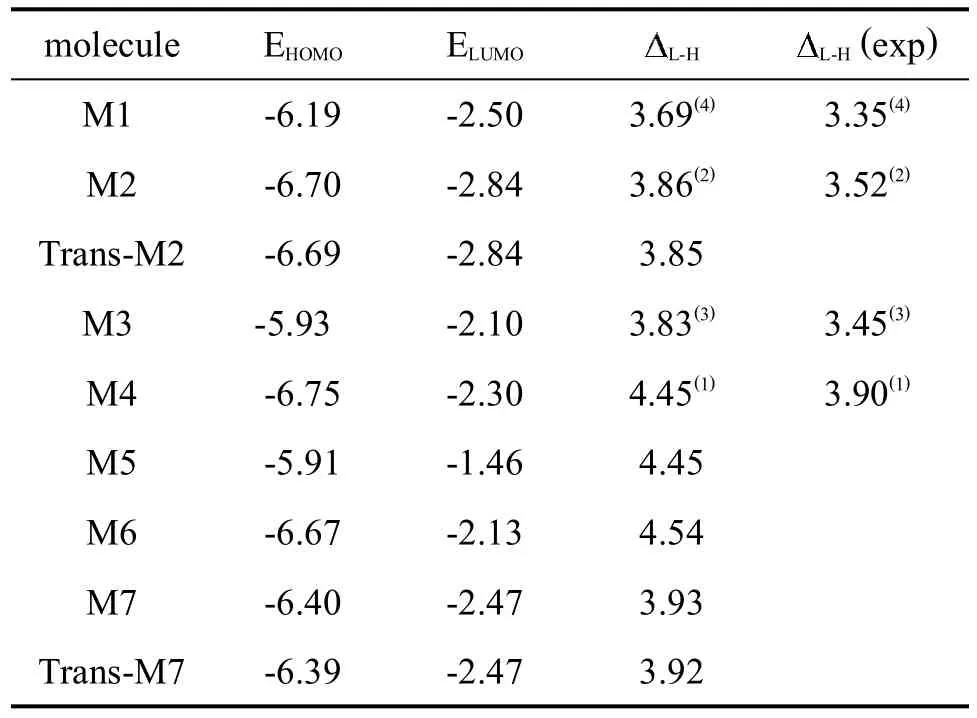
表2 模型分子的前線分子軌道能級
吸電子基使HOMO和LUMO的能級都降低[2,3]。在表2中,同類型分子的LUMO、HOMO值,M1〉M2,M3〉M7,符合這一規律。進一步從分子結構特征出發,將雙噁二唑分子M1與M3、M2與M7比較,可得出分子中四氟苯基對LUMO、HOMO的影響;將雙噁二唑分子M1與M2、單噁二唑分子M5與M6和M4比較,可得出三氟甲基對LUMO、HOMO的影響。用計算數據分析可以得出的結論是,三氟甲基對HOMO的影響要大于四氟苯基,與紫外光譜的結果一致,如在M1與M2的對比中,三氟甲基使M2的HOMO值比M1降低了-0.51(ev),而在M1與M3、M2與 M7的對比中,四氟苯基卻使其只分別降低了-0.26(ev)和-0.30(ev);兩種基團對LUMO的影響相對都要小一些,在 M1與 M2中,M2只降低了-0.340(ev),在M1與M3中,M1降低了-0.40(ev);M2與M7中,M2降低了-0.27(ev)。在單噁二唑分子M5與M6和M4中,三氟甲基對兩種能級的影響都比較明顯,M6和M4的HOMO值比M5分別降低了-0.76和-0.84(ev),LUMO分別為-0.67、-0.62(ev)。上述結果與文獻報道的影響規律[2,3]完全一致。從三氟甲基對單、雙噁二唑的影響看,對單噁二唑的影響要大得多,這可能是因為雙噁二唑分子中共軛程度大,受外界的影響也要小得多的緣故。總之,含有三氟甲基的M2和M7的HOMO軌道的能量都比不含該基團的分子M1和M3的HOMO能量低。根據是否含有三氟甲基和四氟苯基及它們對HOMO的影響大小,還顯示出一定的規律性:M2〈M7〈M1〈M3。但這種吸電子基團造成的結構差異,導致它們的光電性質出現理論與實際有一定差別。有的差別在理論研究中比較明顯[1,2],而在實驗研究中不是十分顯著[26,30]。對于這種理論與實際的差別,如前所述,一般都認為是由于模型分子在實驗中的條件與理論計算時的假定有很大區別而造成的。因為在理論計算時,是將分子假設為單個的氣態分子,而在實際測定時,分子是處于溶液或薄膜中,由此存在著溶劑效應[29,30]或分子間的鏈堆積效應,從而對結果產生影響,但這并不會影響從中得出的有用信息[1]。表3中還給出了具有順反幾何結構的 M2與M2-Trans,M7和 M7-Trans的計算結果,它們的HOMO與LUMO能級和分子總能量基本沒有差別,但同時含有三氟甲基和四氟苯單元的分子的能量要低得多。可以預言,這些分子的熱穩定性應該比較高。
2.3.2 分子軌道成分分析
我們選擇了具有代表性特征的化合物M1、M2、M3、M4,將它們的前線分子軌道圖在圖3中給出。

圖3 化合物M1,M2,M3,M4的前線分子軌道
從圖3可以看出雙噁二唑分子M1、M2、M3的HOMO軌道的主要成分集中在苯環和噁二唑環,而LUMO軌道主要由中間苯環和少量噁二唑環組成,但以苯環為主,端基苯環上僅有少量分布。據此可推斷,中間和兩端苯環上更強或更多吸電子基團的取代同時有利于降低HOMO能級和LUMO能級,中間苯環上的更強或更多吸電子基團的取代則有利于降低LUMO能級。另外,前線軌道中幾乎不含三氟甲基和氟原子成分,但卻使前線軌道能級降低,說明吸電子基團三氟甲基和氟原子具有強烈的誘導效應,這一點與文獻完全一致[2,29]。在M4的分子軌道圖中,軌道成分在HOMO中的分布,苯環上要大一些,噁二唑環上要稍小一些;在LUMO能級上的軌道分布,苯環上所占比例比HOMO則要更大一些,噁二唑環上要更小一些,與文獻基本一致[4],總體上HOMO與LUMO的軌道成分差別不是太大,因此,導致吸電子基團的誘導作用,都使它們的能級大幅降低。這些結論與上面計算出的分子軌道能級高低順序也是一致的。
3 結論
通過運用B3LYP/6-31G(d)方法,計算和優化了一系列含有三氟甲基與氟吸電子基團的噁二唑類衍生物模型分子的光電性能和結構。研究表明,在這一系列的噁二唑類衍生物中,四氟苯基和三氟甲基在理論上都能影響它們的光電性質,但四氟苯基的影響沒有三氟甲基強,主要因素是它的吸電子效應對模型分子的紅外振動、電子吸收光譜和前線軌道能量以及分子能量有規律的影響,得到了與實驗相一致的結論,對今后設計合成該類光電材料具有理論上的指導意義。
[1]Yang L,Liao Y,Feng J-K,et al.Theoretical studies of the modulation of polymer electronicand optical propertiesthrough the introduction of the electron-donation 3,4-ethylenedioxythiophene or electron-accepting pyridine and 1,3,4-oxadiazolemoieties[J].J Phys Chem A,2005,(109):7764-7774.
[2]孟素慈,黃宗浩,徐棟,等.共軛CN與非共軛CF3吸電子取代基對PPV類衍生物分子結構及光電性質的影響[J].化學學報,2004,62(11):1065-1070.
[3]黃宗浩,段雪梅,闞玉和,等.給/吸電子基團取代對PPV類電致發光聚合物光電特性的影響[J].高等學校化學學報, 2002,23(12):2340-2343.
[4]李小兵,王學業,高進偉.電子傳輸材料1,3,4噁二唑衍生物和1,2,4三唑衍生物的DFT研究[J].化學物理學報,2005, 18(6):931-936.
[5]葛夢媛,趙鑫,顧楨燕,等.新型含噁二唑雙Shiff堿的合成及光電性能[J].蘇州科技學院學報(工程技術版),2012,25(1): 43-46.
[6]高健,劉煜,譚華,等.1,2-亞乙基橋練咔唑和芳基噁二唑的雙極傳輸材料的合成及光電性能研究[J].化學學報,2010,68(7):661-666.
[7]王云,李東風,馬興榮,等.新型噁二唑類衍生物的合成及熒光光譜分析[J].化工新型材料,2011,39(5):37-40.
[8]汪新,沈娟,徐洪耀.新型噁二唑電子傳輸材料的分子設計合成、結構與性能關系研究[J].功能材料,2008,39(9):1555-1558.
[9]張小兵,唐本臣,田文晶,等.新型1,3,4-噁二唑衍生物的能帶結構及其對器件性能的影響[J].高等學校化學學報,2007, 28(4):794-797.
[10]張小兵,張子敏,張玉貴,等.新型1,3,4-噁二唑衍生物的合成與性質研究[J].有機化學,2009,29(2):297-301.
[11]AlexanderVGaenko,Ajitha Devarajan,Rostislav ETrifonov,et al.Spectral and Density Functional Studies on the Absorbance and Fluorescence Spectra of 2-R-5-Phenyl-1,3,4-oxadiazoles and Their Conjugate Acids[J].J Phys Chem A, 2006,110(28):8750-8757.
[12]Davut Avci,Yusuf Atalay.Theoretical analysis of vibrational spectra and scaling-factor of 2-aryl-1,3,4-oxadiazole derivatives[J].International Journal of Quantum Chemistry,2009, 109(2):328-341.
[13]Chan L,Yeh H,Che C.Blue Light-emitting devices based on molecular glass materials of tetraphenylsilane compounds [J].Adv Mater,2001,(13):1637-1641.
[14]Cha S W,Choi S-H,Kim K,et al.Synthesis and lumin escence properties of four-armed conjugated structures containing 1,3,4-oxadiazole moieties[J].J Mater Chem,2003, (13):1900-1904.
[15]Ichikawa M,Kawaguchi T,Kobayashi K,et al.Bipyridyl oxadiazoles as efficient and durable electron-transporting and hole-blocking molecular materials[J].J Mater Chem,2006, (16):221-225.
[16]Kamtekar K-T,Wang C-S,Bettington S,Batsanov A-S,et al. New electroluminescent bipolar compounds for balance charge-transport and tuneable colour in organic light emitting diodes:triphenylamine-oxadiazole-fluorene triad molecules [J].J Mater Chem,2006,(16):3823.
[17]Wang C-S,Palsson L,Batsanow A-S,etal.Molecular wire comprising -extended ethynyl-and butadiynyl-2,5-diphenyl-1,3,4-oxadiazole derivatives:Synthesis,redox,structural, and optoelectronic properties[J].J Am Chem Soc,2006, (128):3789-3799.
[18]Hughes G,Bryce M-R.Electron-transporting materials for organic electroluminescent and electrosphorescent devices [J].J Mater Chem,2005,(15):94-107.
[19]Wang C,Jung G-Y,Hua Y,et al.An efficient pyridine and oxadiazole-containing hole-blocking material for organic light-emitting diodes:Synthesis,crystal structure,and device performance[J].J A K Chem Mater,2001,(13):1167-1173.
[20]Heidenhain S-B,Sakamoto Y,Suzuki T,et al.Perfluorinated Oligo(p-Phenylene)s:Efficient n-Type Semiconductors for Organic Light-Emitting Diodes[J].J Am Chem Soc,2000, (122):10240-10241.
[21]Sakamoto Y,Suzuki T,Miura A,et al.Synthesis,Characterization,and Electron-Transport Property of Perfluorinated Phenylene Dendrimers[J].J Am Chem Soc,2000,(122): 1832-1833.
[22]Ohkubo K,Sakamoto Y,Suzuki T,et al.Synthesis,Structure, and Transport Property of Perfluorinated Oligofluorenes[J]. Chem Eur J,2008,(14):4472-4474.
[23]Burke J M,Thomas R-Ll,Collings J-C,et al.Structure and Phase Behavior of a 2:1 Complexbetween Arene-and Fluoroarene-BasedConjugated Rigid Rods[J].Angew Chem,2004, (43):3061-3063.
[24]Delgado M C-R,Pigg K-R,Filho D-A,et al.Impact of Perfluorination on the Charge-Transport Parameters of Oligoacene Crystals[J].J Am Chem Soc,2009,(131):1502-1512.
[25]Martin E,Hughes D L,Hursthouse M B,et al.The synthesis, structure and reactivity of 4-nonafluorobiphenyl complexes [J].J Dalton Tran,2009,1593-1601.
[26]朱萬強,唐國風,勾華.一類含氟芳基噁二唑化合物的合成及其光物理和電化學性質研究[J].化學學報,2007,65(17): 1875-880.
[27]朱萬強,高智席,周光明,等.1,3,4-噁二唑衍生物的合成與表征及吸電子基團對電子光譜的影響([J].西南師范大學學報(自然科學版),2008,33(4):116-120.
[28]Suchi G,Kazuo N.Electronic structures and spectral properties of endohedral fullerenes[J].Coordination Chem Rev, 2005,249:1111-1132.
[29]催明俠,董士紅,王文亮,等.4-(1,2-二苯基)乙烯基-4′-(N, N-二苯基-4-乙烯基苯胺基)聯苯及其二氟取代衍生物的電子結構與光譜性質[J].物理化學學報,2009,(25):157-162.
[30]黃春輝,李富友,黃巖誼.光電功能超薄膜[M].北京:北京大學出版社,2001.238.
(責任編輯:朱 彬)
A Theoretical Study of the Optical Properties of 2,3,5,6-Tetrafluorophenyl and Trifluoromethylphenyl Groups Substituted 1,3,4-Ox adiazole Compounds
ZHU Wan-qiang1,2,YANG Yuan3
(1.Chemistry andChemicalTechnology Institute,Zunyi Normal College,Zunyi 563002,China;2.Laboratory ofUtilization Research onCharacteristic Resources in Qianbei,Zunyi 563002,China;3.College of Chemistry,Guizhou Normal University,Guiyang 550000,China)
A density functional theory(DFT)at a B3LYP/6-31G(d)level was used to optimize the UV-Vis spectra of the 1,3,4-oxadiazoles containing 2,3,5,6-tetrafluorophenyl and trifluoromethylphenyl groups.The results showed that both trifluoromethylphenyl and 2,3,5,6-tetrafluorophenyl groups influenced the optical properties of the oxadiazoles.In comparison with the 1,3,4-oxadiazoles without fluoro group,trifluoromethylphenyl and 2,3,5,6-tetrafluorophenyl groups substituted 1,3,4-oxadiazoles exhibited lower LUMO and HOMO energy levels.Moreover,the influence of trifluoromethylphenyl group was larger than that of 2,3,5,6-fluorophenyl group.As a result,the oxadiazoles containing trifluoromethylphenyland 2,3,5,6-tetrafluorophenyl groups showed ablueshiftintheirUV-Visspectra, which matched well with the previously reported experimental results.
a density functional theory;oxadiazole derivatives;2,3,5,6-tetrafluorophenyl;trifluoromethylphenyl;UV-Vis spectra
0641.12
A
1009-3583(2014)-0062-06
2013-12-24
貴州省科學技術基金資助項目(黔科合J字LKZS[2012]27號);遵義市科技局基金資助項目(遵市科合社字[2009]19號)
朱萬強,男,貴州遵義人,遵義師范學院化學化工學院教授,主要從事無機化學、材料化學等研究。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
