警惕造成“血管衰竭”的一大病因—餐后高血糖
2014-06-01 10:39:53井上輝雄
糖尿病天地(臨床) 2014年6期
井上輝雄 等
臨床綜述
警惕造成“血管衰竭”的一大病因—餐后高血糖
井上輝雄 等
餐后高血糖表現為餐后血糖的高峰,這可以引起內皮細胞功能障礙、炎癥反應和氧化應激,進而導致動脈粥樣硬化的進展和心血管事件的發生。已有資料顯示餐后高血糖甚至糖耐量減低即可使動脈粥樣硬化和心血管事件更易于發生。有證據顯示餐后高血糖可獨立預測心血管事件的發生,而空腹高血糖則不能。我們提出“血管衰竭(vascular failure)”的概念,用以概括血管功能障礙的所有表現,包括從危險因素到動 脈粥樣硬化性疾病晚期。因此餐后高血糖是造成血管衰竭的一種非常重要的病理生理狀態。因此,控制餐后高血糖應該作為預防血管衰竭一個潛在的目標而成為未來臨床研究的焦點。
引言
2型糖尿病可使冠狀動脈粥樣硬化及腦血管疾病的發生風險顯著升高。另外,有證據表明,餐后血糖異常狀態尤其是餐后高血糖是動脈粥樣硬化的獨立危險因素。最近的流行病學調查顯示,餐后高血糖是心血管疾病的獨立危險因素,它對心血管疾病的影響比空腹高血糖更大。
動脈粥樣硬化是血管壁對多種原因造成慢性損傷的反應引起的一種進展性疾病,最終導致動脈粥樣硬化或纖維斑塊的形成。通常認為內皮細胞功能障礙是動脈粥樣硬化的起始階段。除此以外,動脈粥樣硬化進程的早期階段即存在血管壁平滑肌細胞功能障礙、代謝異常,包括炎癥反應、氧化應激和神經激素失衡等。我們最近提出“血管衰竭(vascular failure)”的新概念,用以代表上述各種血管異常的整體情況。Schwartz等人曾使用“血管衰竭”一詞表示血管重塑反應喪失。與之不同,我們把“血管衰竭”定義為一種血管功能障礙的整體性的綜合征,涵蓋從危險因素到動脈粥樣硬化晚期的動脈狹窄,直至最終導致血管壁鈣化,以及斑塊破裂或血栓栓塞等嚴重血管事件(圖1)。
餐后高血糖的病理生理過程:血糖高峰引起氧化應激,生成可溶性糖基化終末產物(AGEs)及脂質過氧化物,并作為上游激酶的關鍵性活化因子,導致內皮細胞功能障礙及炎癥基因表達(圖2)。餐后高血糖在血管衰竭發病過程中是一種重要的基本功能紊亂,本文回顧餐后高血糖在其中的作用。
最近有證據表明,有癥狀的心血管疾病患者中將近2/3存在血糖穩態失衡。這些患者絕大部分空腹血糖水平未見升高,而在進食后或口服葡萄糖耐量試驗時發現餐后血糖水平升高。葡萄糖不耐受通常由75克葡萄糖耐量試驗方可確定。葡萄糖負荷后2小時血糖水平140~200毫克/分升可診斷為糖耐量減低(IGT)或稱糖尿病前期,若超過200毫克/分升即可診斷2型糖尿病。
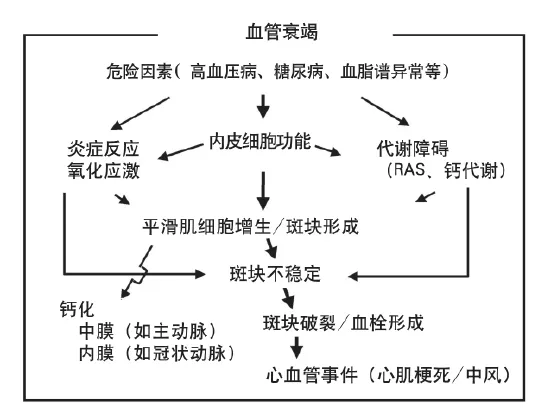
圖1 血管衰竭
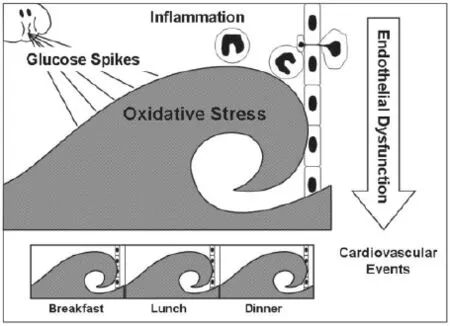
圖2 餐后高血糖的病理生理
即使在通過糖化血紅蛋白A1c(HbA1c)和空腹血糖水平評估認定血糖控制良好的糖尿病患者中,餐后高血糖也經常發生。人群研究表明,空腹血糖水平低至90毫克/分升仍可能伴隨著餐后2小時血糖水平>200毫克/分升。在2型糖尿病的早期階段,即使空腹血糖與HbA1c在正常范圍,餐后高血糖同樣會引起微血管并發癥以及心肌梗死、卒中等大血管并發癥。新近資料顯示,即使IGT也可使動脈粥樣硬化和心血管事件更易于發生。有證據表明,餐后高血糖可獨立預測心血管事件,而空腹高血糖不能。例如,Funagata糖尿病研究(Funagata Diabetes Study)結果表明,與空腹血糖和糖化血紅蛋白相比,負荷后1小時或2小時血糖水平能更好的預測心血管風險,而糖尿病流行病學:歐洲糖尿病診斷標準合作分析(Diabetic Epidemiology:Collaborative Analysis of Diagnosis Criteria in Europe,DECODE)顯示,負荷后2小時血糖水平與心血管死亡風險間存在直接的連續級配關系。即使在糖耐量正常(負荷后血糖<140毫克/分升)患者中,負荷后血糖水平也與心血管死亡及全因死亡風險相關。負荷后血糖達到80毫克/分升后心血管風險即開始升高,達到140毫克/分升(傳統歸為IGT或糖尿病前期)時,心血管風險升高58%。
內皮細胞功能障礙是動脈粥樣硬化初始階段的觀點現已得到廣泛的認同。內皮細胞功能障礙時,血管內皮細胞來源的舒張因子尤其是L-精氨酸在內皮一氧化氮合酶(eNOS)作用下生成的一氧化氮(NO)生成減少,生物利用度降低。NO生物利用度減少導致依賴內皮細胞的血管舒張作用損害,這就是內皮細胞功能障礙的功能性表現。另一方面,它還包含一個特定的“內皮激活”的狀態,表現為促炎、增生及血栓前狀態,這種狀態對動脈粥樣化形成的各個階段均有促進作用。鑒于內皮細胞功能障礙和動脈粥樣硬化之間的這種關系,內皮細胞功能狀態很可能反映了個體發生動脈粥樣硬化性疾病的傾向性。因此,內皮細胞功能障礙可以作為早期血管衰竭的標志。
不僅糖尿病時內皮細胞功能損害,IGT時也是同樣。我們觀察到與糖耐量正常的受試者相比,IGT患者血流介導的血管舒張(FMD)減弱。血糖迅速升高可影響內皮細胞功能,體內、體外實驗均證實高血糖具有這種直接的作用。體外實驗中僅通過簡單的將標本暴露于高濃度的葡萄糖環境中,就發現乙酰膽堿介導的依賴內皮細胞的血管舒張作用減弱,并表現出濃度依賴性。體內實驗同樣顯示血糖高峰誘導內皮細胞功能障礙,Williams等人利用FMD技術發現,動脈灌注50%右旋葡萄糖引起的急性高血糖,可使健康受試者依賴內皮細胞的血管舒張作用減弱。這個結果表明血糖水平升高是糖尿病相關的內皮細胞功能障礙的一個主要原因。Shige等人也發現2型糖尿病患者進食高脂肪或高蔗糖食物后FMD減弱。上述研究中餐后FMD水平及變化程度均與餐后血糖水平變化顯著相關,因此可以確認餐后高血糖是FMD減弱的一個決定性因素。
高血糖的這些作用可能與NO的生成和/或生物利用度減少有關,高血糖誘導的內皮細胞功能障礙可能被精氨酸的生成增加所拮抗。NO的生成與生物利用之間的相互關系尚未明確,尚未明確高血糖是否能減少NO的生成,或者,更有可能的是,NO生成的增加引起它的抑制物超氧陰離子的顯著增加,進而導致NO生物利用度的降低。而且已經發現,IGT患者進行OGTT試驗時FMD的迅速降低與2小時血糖水平相關。
最近的資料表明,AGEs可以影響內皮細胞功能障礙的進展。AGEs是一組具高度異質性的化合物,最具代表性的是羧甲基賴氨酸(CML)。飲食是外源性AGEs的一個主要來源,食物AGEs含量與食品營養成分含量高度相關,另外也與烹飪的方法、溫度及持續時間有關。飲食來源的AGEs大約10%迅速吸收,部分保留在體內,結合并激活AGE受體,進而起到多種病理作用。AGE前體如甲基乙二醛(MG)也可以激活AGE受體,有體內研究顯示高血糖與內生MG合成增加相平行。進餐后,吸收和內生的AGEs和MG協同作用,直接清除NO,促進氧化應激,進而降低血管功能。
餐后高血糖與炎癥反應
現已公認炎癥反應是血管衰竭發病機制中的一個主要因素。長期以來,一直通過炎癥標志物如高敏C-反應蛋白(hsCRP)來評價糖尿病時的炎癥反應。炎癥細胞和介質在炎癥過程中起著至關重要的作用。動脈粥樣硬化起始及進展時,白細胞如單核細胞和T-淋巴細胞在動脈血管壁內皮浸潤,內皮與白細胞之間通過黏附分子調節其相互作用。在各種黏附分子中,細胞間黏附分子-1(ICAM-1)和血管細胞黏附分子-1(VCAM-1)特別引人注意。已經證實血管疾病和糖尿病(不論其是否有血管疾病)時這些分子的循環形式增加。我們發現與健康受試者相比,IGT患者的循環ICAM-1水平已有升高,但VCAM-1未見這種變化(圖3)。此外已經證實在正常和糖尿病受試者中,急性高血糖均可以增加循環ICAM-1水平,進而激活動脈粥樣硬化進程的起始階段。現已有證據證實高葡萄糖鉗夾或進餐后引起的急性高血糖均可促進血漿白介素-6、腫瘤壞死因子-α及白介素-18生成增加,因此,現已公認動脈粥樣硬化是一種炎性疾病,即使在合并糖尿病時也是這樣。
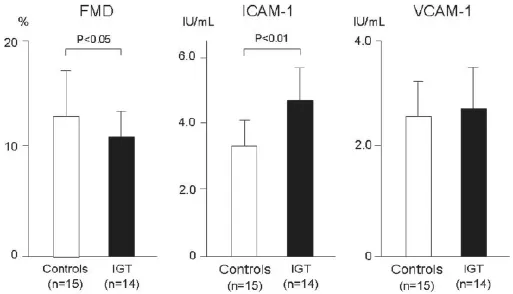
圖3 糖耐量減低(IGT)患者與正常糖耐量受試者(對照組)內皮細胞功能與炎癥反應標志物
先前的研究主要集中在單核細胞和T-淋巴細胞方面,因為它們是參與動脈粥樣硬化的炎癥細胞的主要類型,但越來越多的證據表明,中性粒細胞活化在缺血性心血管疾病的炎癥進程,特別是在急性炎癥應答中,也扮演了重要的角色。此外一項對冠狀動脈疾病和腦血管疾病高危患者的大規模隊列研究顯示,中性粒細胞計數在白細胞分類中是未來心血管事件最好的預測因子。外周中性粒細胞是活性氧最重要的來源。中性粒細胞的激活可顯著影響2型糖尿病的氧化應激和炎癥反應,繼而導致血管病及動脈粥樣硬化。β2-整合素Mac-1(CD11b/ CD18)是一種對中性粒細胞及單核細胞緊密黏附于血管內皮細胞起到至關重要的作用的黏附分子。Mac-1與血管內皮表面ICAM-1結合,并通過結合纖維蛋白原或血小板受體如糖蛋白(GP)Ibα、ICAM-2等而與血小板結合。臨床研究表明,Mac-1在冠狀動脈血管成形術后粘附在損傷血管處的中性粒細胞表面激活和上調。2型糖尿病患者中也發現中性粒細胞表面Mac-1上調。另外有報道體外實驗中,激動劑誘導糖尿病患者中性粒細胞表面Mac-1上調增強。此前我們曾對未診斷糖尿病、空腹血糖<126毫克/分升的患者,在口服75克葡萄糖負荷120分鐘及以前,使用流式細胞術觀察孤立中性粒細胞表面Mac-1的表達情況。結果顯示,與基線相比,餐后高血糖(120分鐘血糖水平≥200毫克/分升)患者葡萄糖負荷120分鐘后,Mac-1甚至在未激活的中性粒細胞表面上調。此外與基線相比,餐后高血糖及IGT(120分鐘血糖水平≥140毫克/分升)患者fMLP介導的Mac-1上調在120分鐘時顯著增強,而這些變化在正常糖耐量(120分鐘血糖水平<140毫克/分升)患者中并不明顯(圖4)。這些結果表明,血糖急性升高可能使餐后高血糖和IGT患者中性粒細胞從非活化形式改變為潛在激活狀態。
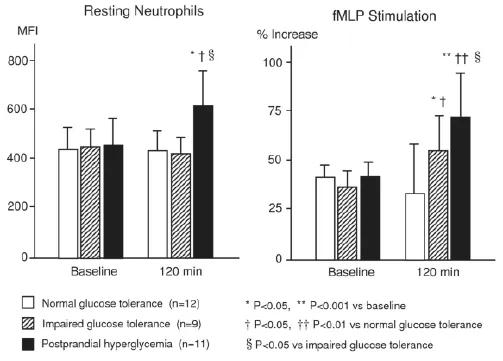
圖4 口服75克葡萄糖負荷后120分鐘及口服前β2-整合素Mac-1在孤立中性粒細胞表面的表達
餐后高血糖對氧化應激的影響
氧化應激是細胞的異常氧化還原狀態,在血管衰竭尤其是血管內皮細胞功能障礙的發病過程中起到關鍵的作用。超氧陰離子是氧分子經一價還原形成的,包括黃嘌呤氧化酶、NADH/NADPH氧化酶、脂氧合酶、NOS在內的多種酶類參與這個過程,線粒體是體內超氧陰離子生成的主要場所。超氧陰離子自發的或是經酶催化的歧化反應還原生成過氧化氫,后者經金屬(鐵或銅)催化轉化,生成具有很強毒性的羥基自由基。近來研究顯示高血糖介導超氧陰離子在線粒體電子轉運鏈的作用下大量合成超氧陰離子。超氧化物的過量生成造成eNOS與誘導型NOS(iNOS)之間的非耦合狀態,導致NO合成增加,進而促進強氧化劑——過氧亞硝基的生成,最終損害DNA。DNA損傷后方可激活細胞核內多聚ADP-核糖聚合酶,繼而消耗細胞內的底物NAD+,使糖酵解、電子傳遞及三磷酸腺苷的生成速度減慢,進而導致磷酸甘油醛脫氫酶活性降低,使二磷酸腺苷核糖化過程減慢。上述這些過程均造成糖尿病血管急性內皮細胞功能障礙,極易引起心血管疾病。現已同時有直接和間接兩方面的證據證明,急性高血糖通過氧化應激產物促進IGT受試者心血管疾病的進展。直接證據是基于餐后高血糖氧化應激標志物的作用,如硝基酪氨酸和8-異前列腺素F2α(8-iso-PGF2α)。
氧自由基與一氧化氮反應生成過氧亞硝基,后者一種強氧化劑,可以通過硝基類似物中介使側鏈與肽鍵消除形成羰基從而直接氧化蛋白質、脂類及DNA。過氧亞硝基與酪氨酸殘基之間存在親和力,相互反應生成硝基酪氨酸。一些研究結果表明,高血糖可直接促進硝基酪氨酸的過量生成,例如,猴高血糖時的動脈壁上、健康受試者高血糖鉗夾或OGTT時以及糖尿病患者餐后高血糖情況加重時的血漿中均檢測到硝基酪氨酸的生成,而且發現硝基酪氨酸的生成依賴于血糖水平。有研究發現大鼠心臟灌注中高血糖伴隨著硝基酪氨酸沉積,這似乎與iNOS過度表達使NO與超氧陰離子生成不均衡有關。已經證實硝基酪氨酸的生成可導致健康受試者和灌注心臟冠狀動脈的內皮細胞功能障礙。已經證實硝基酪氨酸可直接損傷內皮細胞,因此這種作用并不會令人感到驚奇。
特殊的異前列腺素異構體例如尿液中的8-iso-PGF2α已經公認是氧化應激的標志物。異前列腺素均由自由基介導的花生四烯酸氧化生成,后者廣泛分布于細胞膜上,尿液異前列腺素測定能更好的反映整個機體的氧化應激狀況。一些研究顯示高血糖與8-iso-PGF2α的生成及在尿液中的排泄速度加快有關。據報道,與對應年齡的健康受試者相比,2型糖尿病患者尿液8-iso-PGF2α排泄率顯著增加,而且發現血糖水平與尿中8-iso-PGF2α存在顯著相關性,提示氧化應激的增加至少可能部分上與糖尿病控制的決定性因素存在相關性。另有體外高血糖條件下培養豬血管平滑肌細胞發現8-iso-PGF2α的合成及釋放增加,與上述結果相符合。在最近的一項研究中,Monnier等證實尿液8-iso-PGF2α排泄率與通過平均血糖漂移幅度(MAGE)評估的血糖變異性之間有很強的正相關關系,而且與平均餐后血糖升高間在統計學上具有顯著相關性,雖然并非強相關。這些發現表明,與急性餐后血糖高峰相比,急性血糖漂移對氧化應激觸發作用的影響更加廣泛,因而更應該被納入血糖紊亂的范疇之內。因此,餐后血糖高峰是“危險的浪潮”,這種認識應該擴展到血糖受間斷進食的影響而在平均值上(餐后)下(進餐間隔期間)急性波動。
治療例證
一些臨床試驗表明,特定的藥物可以降低餐后血糖漂移對總體血糖控制的影響。但到目前為止,尚無評價改善餐后高血糖對糖尿病長期預后影響的前瞻性臨床研究。根據餐后血糖對總體血糖控制影響的新近資料,專注于餐后血糖的治療手段很可能使患者長期受益。傳統的糖尿病藥物如胰島素、磺脲類藥物等主要降低空腹血糖,而對控制餐后高血糖效果較差。磺脲類藥物作用于β細胞鉀通道而直接促進胰島素分泌,但也可能對空腹和餐后血糖均有影響。然而大多數磺脲類藥物的藥物代謝動力學情況并不針對胰島素急性分泌,因此他們不能改善胰島素第一時相分泌異常。另一方面,一些藥物的藥代動力學及作用機制特別針對餐后高血糖。這樣的藥物包括格列奈類、α-糖苷酶抑制劑和噻唑烷二酮類藥物。
現今非磺脲類促泌劑格列奈類藥物包括瑞格列奈、那格列奈和米格列奈,用以恢復第一時相胰島素分泌。已經證實單劑量的那格列奈通過降低2型糖尿病負荷后血糖而緩解餐后內皮細胞功能障礙。在一項針對糖尿病的隨機試驗中,磺脲類藥物格列本脲降低空腹血糖水平更有效,而瑞格列奈能更大程度的降低餐后血糖水平。兩組HbA1c降低的幅度相同,但瑞格列奈可使52%的患者的頸動脈內膜中層厚度得到恢復,而格列本脲組這一比例只有18%,而且動脈粥樣硬化的恢復程度與餐后高血糖的降低幅度成正比。
α-糖苷酶抑制劑通過阻斷α-糖苷酶而減慢碳水化合物包括淀粉和二糖的分解。糖尿病及葡萄糖耐受不良的患者應用α-糖苷酶抑制劑可使負荷后血糖峰值降低30~70毫克/分升、糖化血紅蛋白降低約0.7%,而對空腹血糖水平的影響微乎其微。長期接受阿卡波糖治療可能通過改善胰島素敏感性而降低甘油三酯和乳糜微粒的水平。最近的一項研究表明,即使是單次450卡路里的混合飲食就可以立即造成接受飲食治療的糖尿病患者內皮細胞功能嚴重的破壞。對同樣的受試者預先給予單劑量的阿卡波糖就可以顯著降低餐后高血糖,改善進食后的內皮細胞功能障礙。預防非胰島素依賴型糖尿病(STOP-NIDDM)國際研究共納入1429例糖耐量減低的受試者,隨機分為兩組,每日三次隨餐服用100毫克阿卡波糖或安慰劑。3.3年后,阿卡波糖使研究的主要終點(新發糖尿病)顯著降低25%,并顯著降低任何心血管事件發生率達49%、心肌梗死發生率降低91%。阿卡波糖也可延緩頸動脈粥樣硬化的進展,減少新發高血壓達34%,這些結果說明餐后代謝異常可能參與高血壓的發病。一個包括7項長期研究的大型回顧性薈萃分析顯示,阿卡波糖可使2型糖尿病心臟事件的風險降低35%,心肌梗死的風險降低64%。此前我們也發現經過3個月的阿卡波糖治療后FMD增加,hsCRP及VCAM-1水平降低(圖5)。這表明阿卡波糖可以改善餐后代謝障礙,改善內皮細胞功能和炎癥狀態,可能延緩動脈粥樣硬化的進展。此外,在最近的一項針對輕癥糖尿病的研究中,我們用飲食耐量試驗(meal tolerance testing)比較一種新的α-糖苷酶抑制劑——米格列醇與格列奈類——米格列奈對餐后血糖和胰島素代謝的影響,同時評估其他的葡萄糖代謝相關指標、動脈粥樣硬化指標以及腎功能情況。結果表明,盡管兩種藥物引起的餐后胰島素分泌模式不同,服用3個月后餐后高血糖的改善情況相近(圖6A),米格列醇組1,5-脫水山梨醇水平顯著高于米格列奈組;米格列醇組穩態模型胰島素抵抗指數和尿白蛋白排泄率顯著降低,米格列奈組則不明顯;米格列醇組血清胱抑素C水平無改變,米格列奈組則升高;米格列醇組hsCRP水平顯著降低(圖6B)、血清脂聯素水平顯著升高,而米格列奈組則沒有顯著改變。上述結果表明米格列醇有抗炎和腎臟保護作用,這可能與改善胰島素抵抗有關。
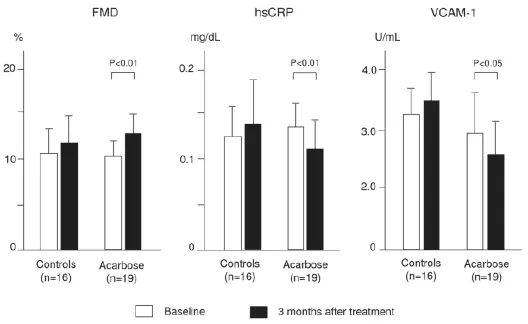
圖5 阿卡波糖對輕癥糖尿病患者血管內皮細胞功能和炎癥狀態的影響
噻唑烷二酮即過氧化物酶體增殖物激活受體γ(PPAR-γ)激動劑類藥物如曲格列酮、羅格列酮、吡格列酮等,可改善胰島素敏感性,而對胰島素分泌的影響通常可以不予考慮。然而經由曲格列酮證實,這類化合物應用于糖耐量減低受試者時,可以促進葡萄糖刺激引起的胰島素分泌,改善餐后高血糖。PPAR-γ激動劑顯示出抗炎及通過調節細胞氧化還原狀態改善內皮細胞功能的作用。現已證實噻唑烷二酮類藥物能降低hsCRP等炎癥標志物水平,提高肱動脈的FMD。一項納入5238名2型糖尿病合并大血管疾病患者的大樣本的隨機臨床試驗證實,與安慰劑相比,吡格列酮與降糖藥物及其他藥物聯合使用后可減少心血管不良轉歸。也有報道與格列美脲相比,吡格列酮可延緩頸動脈內膜中層厚度進展,及通過血管內超聲成像評估的冠狀動脈粥樣硬化的進展。
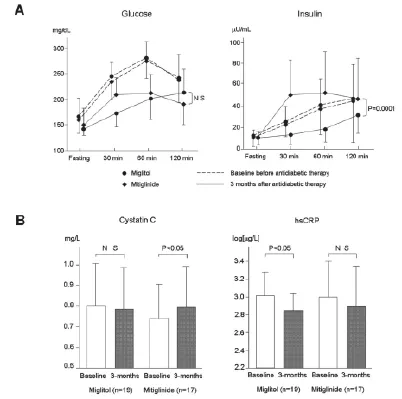
圖6 米格列醇與米格列奈對輕癥糖尿病患者飲食耐量試驗中餐后血糖及胰島素代謝效用對比
最近一些能帶來臨床獲益的新型降糖藥被批準上市,包括注射用藥胰高血糖素樣肽-1(GLP-1)受體激動劑及口服藥物二肽基肽酶-4(DPP-4)抑制劑。GLP-1受體激動劑如艾塞那肽能促進營養物質引起的胰島素分泌,減少胰高血糖素的不當分泌,同時延緩胃排空,控制食欲。這些藥物在堅持減肥時發生低血糖的風險很低。DPP-4抑制劑沙格列汀及維格列汀,對體重通常無影響,胃腸道副作用不如GLP-1受體激動劑嚴重。無論是單獨應用還是與格列本脲或吡格列酮聯合應用,此類藥物表現出良好的耐受性,而且不發生低血糖;與格列本脲或吡格列酮聯合應用可改善2型糖尿病患者的餐后血糖,而對這些藥物的藥代動力學無顯著影響。
結論
餐后高血糖期間,血糖的高峰可導致內皮細胞功能障礙、炎癥反應及氧化應激,進而導致動脈粥樣硬化的進展及心血管事件的發生。流行病學與發病機制研究的資料均表明,餐后高血糖甚至IGT即可影響動脈粥樣硬化性疾病的發生發展,有初步證據表明控制餐后高血糖可以減少IGT人群心肌梗死的發病率。因此,餐后高血糖是導致血管衰竭的一種極為重要的病理生理狀態。相應地,控制餐后高血糖應該作為臨床隨訪時預防血管衰竭的潛在目標。
(林樂乙 編譯遼寧中醫藥大學附屬第二醫院)
10.3969/j.issn.1672-7851.2014.06.008
日本佐賀大學醫學院心血管與腎臟學系
猜你喜歡
中老年保健(2022年5期)2022-08-24 02:35:42
中老年保健(2022年1期)2022-08-17 06:14:56
保健醫苑(2022年6期)2022-07-08 01:26:34
家庭科學·新健康(2022年3期)2022-05-10 00:32:13
中老年保健(2021年5期)2021-08-24 07:07:20
中老年保健(2021年9期)2021-08-24 03:51:04
中老年保健(2021年7期)2021-08-22 07:42:16
中老年保健(2021年11期)2021-08-22 03:15:16
家庭醫學(下半月)(2020年1期)2020-05-11 02:05:44
媽媽寶寶(2017年3期)2017-02-21 01:22:30
