大鼠Pten和Runx3基因5'端非編碼區序列的生物信息學分析
2014-06-14 09:48:02楊晉翔賀梅娟彭繼升賈雪晴
山東醫藥 2014年12期
關鍵詞:胃癌
安 靜,楊晉翔,賀梅娟,彭繼升,魏 玥,賈雪晴
(1北京中醫藥大學,北京100029;2北京中醫藥大學第三附屬醫院;3中華中醫藥學會)
Pten基因作為第一個被發現的具有雙重特異性磷酸酶活性的抑癌基因,在細胞生長發育、凋亡、移動、信號傳遞等方面發揮重要的調控作用,它的正常表達可抑制腫瘤細胞生長,促進細胞凋亡,參與細胞周期的調控以及抑制腫瘤的轉移。研究發現,Pten基因啟動子的甲基化與胃癌的發生有關,Pten甲基化是胃癌患者診斷及預后的候選標志物之一[1]。近幾年發現,Runx3是胃癌特異性很高的一個抑癌基因[2]。Runx3在胃黏膜上皮細胞生長及分化調控等方面發揮重要作用[3],其有望成為胃癌診斷的一個特異性生物學標志物和基因治療靶點。2013年10~12月,我們采用DNA序列分析法對大鼠Pten和 Runx3基因5'端非編碼區(5'-UTR)序列的CpG島、啟動子及其轉錄因子結合位點進行預測,旨在為下一步大鼠Pten和Runx3基因甲基化實驗上下游引物設計奠定基礎。
1 材料與方法
1.1 大鼠Pten和Runx3基因全長序列、轉錄本信息獲得 通過美國國立生物技術信息中心(http://www.ncbi.nlm.nih.gov)搜索 Genebank 獲得大鼠Pten、Runx3基因的登陸號、全長序列及轉錄本信息。
1.2 大鼠Runx3和Pten基因外顯子、內顯子及上游5'-UTR信息獲得 通過序列比對,利用美國國立生物技術信息中心Blast中的可讀框(ORF)(http://www.ncbi.nlm.nih.gov/gorf/gorf.html)查找分析確定外顯子和內顯子及5'-UTR,也可通過Ensemble查找外顯子、內顯子及5'-UTR,取翻譯起始點(ATG)前2 kb、后1 kb的區域序列做后續分析。
1.3 大鼠Pten和Runx3基因5'-UTR上游CpG島、啟動子區域及其轉錄結合位點預測 采用序列分析。
1.3.1 CpG島分析 應用開放CpG island searcher軟件(http://cpgislands.usc.edu/)、在線 CpGPlot工具(http://www.ebi.ac.uk/Tools/emboss/)、Methprimer2.0 工具(http://www.urogene.org/methprimer/)預測CpG島。①Pten基因CpG島分析:CpG island searcher設定條件:選擇低限%GC(G、C百分比)=50,CpG島實測值/預期值(ObsCpG/ExpCpG)=0.65,長度(Length)=200,距離(Distance)=100。CpGPlot設定條件:實測值/預期值(Obs/Exp)>0.60,Percent C+Percent G >50.00,Length > 200。Methprimer2.0設定條件:CpG島長度(Island size)>200,GC Percent>50.0,Obs/Exp >0.60。②Runx3基因CpG島分析:CpG island searcher設定條件:選擇低限%GC=50,ObsCpG/ExpCpG=0.65,Length=200,Distance=100。CpGPlot設定條件:Obs/Exp >0.60,Percent C+Percent G > 50.00,Length > 200。Methprimer2.0設定條件:Island size>200,GC Percent>50.0。
1.3.2 啟動子信息預測 應用開放軟件NNPP工具(http://www.fruitfly.org/seq_tools/promoter.html)、Promoter scan 工具(http://www-bimas.cit.nih.gov/molbio/proscan/)、FirstEF 工 具 (http://rulai.cshl.org/tools/FirstEF)預測啟動子。
1.3.3 啟動子區域轉錄結合位點預測 應用開放在線工具/軟件 Matlspector(http://www.genomatix.de/)、Match(http://www.gene-regulation.com)和Consite(http://consite.genereg.net)預測啟動子區域轉錄結合位點。
2 結果
2.1 大鼠Pten和Runx3基因全長序列及其轉錄本序列 大鼠Pten基因位于染色體1q41~q43,全長65 kb,登陸號:50557;有 3個轉錄本:AF455569、AF017185、NM_031606,均為 1 212 bp。大鼠 Runx3基因位于染色體5q36,全長73 kb,登陸號:156726;有2 個轉錄本:AF421886.1、NM_130425.1,均為1 230 bp。
2.2 大鼠Pten和Runx3基因外顯子、內顯子及上游5'-UTR信息 通過BLAST序列比對,發現所報道的Runx3和Pten基因其轉錄本5'-UTR長度均相等,且轉錄本序列高度一致。通過Ensemble查找外顯子和內顯子及5'-UTR顯示,Runx3基因第1外顯子(起始外顯子)長度為285 bp,Pten基因第1外顯子(起始外顯子)長度為79 bp。兩個基因取5'端翻譯起始密碼子(ATG)前2 kb、后1 kb序列進行后續分析。
2.3 Pten基因和Runx3基因CpG島的分析結果
2.3.1 Pten基因 CpG island searcher軟件分析結果:CpG島(-1 952~ -70),ObsCpG/ExpCpG=0.808,Length=1 883。CpGPlot工具分析結果:CpG島1(-1 872~ -1 599),274 bp;CpG 島 2(-1 493~-144),1 290 bp。Methprimer2.0 工具預測結果:CpG島1(-1 872~ -1 599),274 bp;CpG 島2(-1 493~-144),1 290 bp 。
2.3.2 Runx3基因 CpG island searcher軟件分析結果,見表 1;CpGPlot工具分析結果,見表 2。Methprimer2.0工具分析結果與CpGPlot工具分析結果相同。
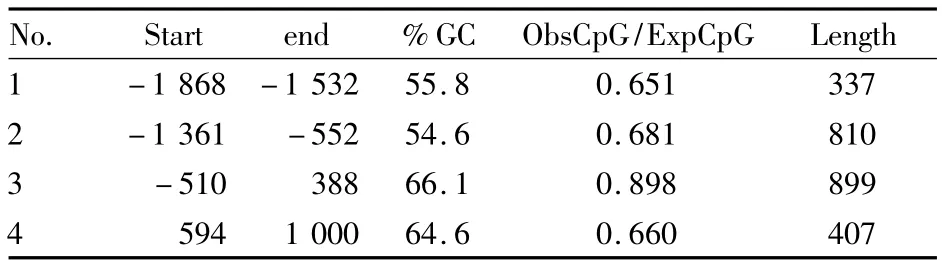
表1 Runx3基因CpG island searcher軟件分析結果
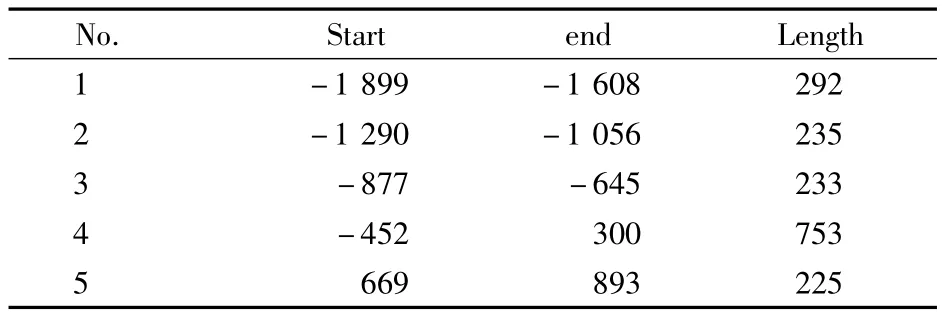
表2 Runx3基因CpGPlot工具分析結果
2.4 Pten基因和 Runx3基因啟動子預測結果NNPP工具預測結果,見表3。Pten基因FirstEF工具顯示CpG島的-1 253~-684 bp是1個啟動子序列,第1外顯子序列為-753~691 bp;Runx3基因FirstEF工具序列-690~-121 bp是1個啟動子序列,第1外顯子序列為-108~41 bp。Promoter scan工具預測結果,見表4。
2.5 轉錄因子結合位點 Pten基因轉錄因子結合位點分析軟件分別顯示CpG島啟動子區上有100、68和107個轉錄因子結合位點,包含有109種轉錄因子;在這109種轉錄因子中,被≥2種軟件共同預測有結合位點的轉錄因子有6種。Runx3基因轉錄因子結合位點分析軟件分別顯示,CpG島啟動子區上有138、71和97個轉錄因子結合位點,包含有94種轉錄因子;在這94種轉錄因子中,被≥2種軟件共同預測有結合位點的轉錄因子有9種。見表5。

表3 Pten基因和Runx3基因NNPP工具啟動子預測結果
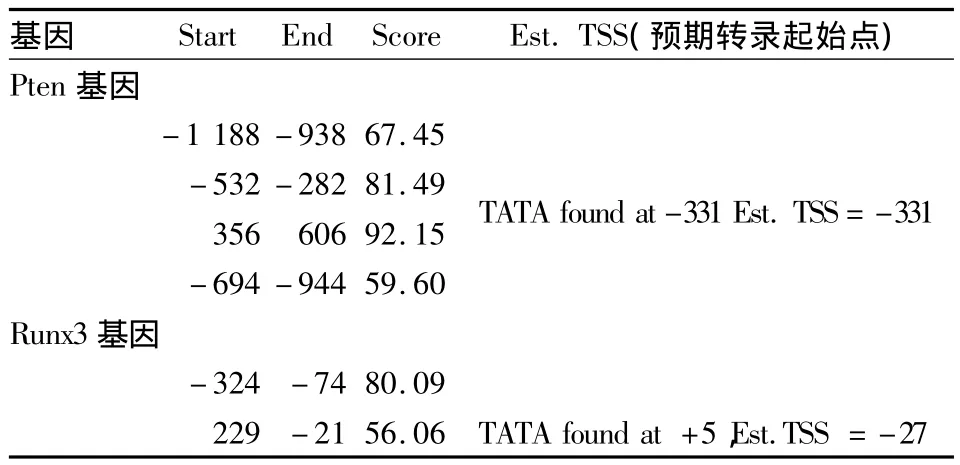
表4 Pten基因和Runx3基因Promoter scan工具啟動子預測結果
3 討論
有研究發現,在多種腫瘤組織或惡性細胞系中存在Pten基因低表達或缺失,并且大部分與其啟動子區甲基化相關。目前認為,突變、雜和性缺失(LOH)及異常甲基化是導致Pten基因失活的主要機制。Runx3在正常胃黏膜上皮細胞廣泛表達,對胃癌高發區大樣本人群的研究證明,Runx3蛋白表達與胃黏膜病變的嚴重程度呈明顯負相關[4]。胃癌組織中Runx3 mRNA和蛋白的表達明顯低于相應正常組織,在不同進展和分化程度的胃癌組織中,Runx3表達也明顯下調,表明Runx3基因與胃癌的進展密切相關[5]。目前發現有多種機制,包括LOH、高甲基化和點突變等參與了Runx3基因在胃癌中的表達缺失或下調,其中Runx3啟動子區域CpG島的甲基化是導致其在胃癌中失活的主要機制。
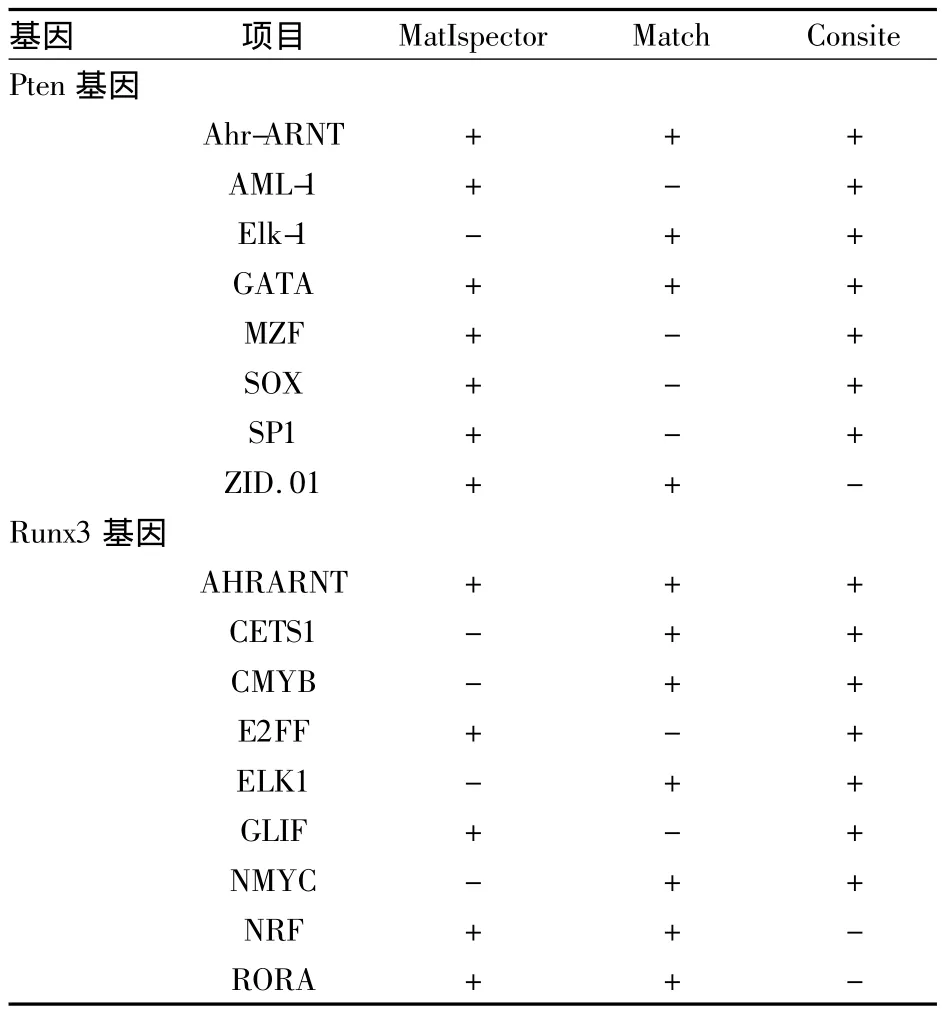
表5 Pten基因和Runx3基因轉錄因子結合位點分析預測結果
本研究發現,大鼠Pten基因和Runx3基因的轉錄本5'端及其上游序列完全位于CpG島內,表明該基因屬于CpG島關聯基因,適合利用FirstEF進行第1外顯子分析。同時,對于CpG相關基因FirstEF軟件對啟動子和第1外顯子預測的敏感性與特異性均高達90%以上。所以,初步確定大鼠Pten基因和Runx3基因核心啟動子可能分別位于-1 253~-684 bp和-690~-121 bp,為進一步基因調控甲基化實驗驗證奠定了基礎。
近幾年,隨著實驗技術的發展,基因組DNA和蛋白質測序數據總量正在以指數倍的速度增長。如何挖掘利用現有海量數據來預測有關基因的CpG島、啟動子和轉錄因子結合位點是生物信息學研究熱點。
CpG島指一段200 bp或更長序列的DNA序列,G+C含量較高[6]。CpG島通常位于基因的5'端,尤其是啟動子和第1外顯子附近,人類基因概率達60%~80%,也是發生甲基化的區域[7]。本研究中所應用軟件在ATG上游均檢測到典型的CpG島結構,相關基因CpG島與啟動子和第1外顯子高度重合,因此查找CpG島能對基因啟動子預測有重要意義。
預測有關基因的啟動子及轉錄因子結合位點有許多算法,而且還有很多軟件和工具提供在線分析。本研究預測啟動子分別基于信號的預測方法和基于內容的預測方法。Promtor Scan屬于基于信號方法識別核心啟動子TATA-box、CAAT-box和TSS等一些重要的啟動子調控元件;NNPP工具預測啟動子是基于信號內容的神經網絡識別TATA-box、CAAT-box、加帽位點和GC框的位置和距離,使用4個結構相同的人工神經網絡分別識別以上4種元件來區別啟動子和非啟動子。另外,基于相關基因CpG島高關聯性,還可采用CpG島關聯性的預測方法。FirstEF是基于二次判別析分析技術[8],先判斷是否為CpG島相關,然后通過搜索第1外顯子數據庫,識別RNA聚合酶酶切點,最后結合CpG島信息,確定啟動子區。因使用了3種不同的二次判別函數,使該方法預測含CpG島的啟動子的敏感性和特異性都高于0.90[9],預測不含CpG島的啟動子的精確性相對略低。盡管目前預測工具很多,但對啟動子識別精度都不高[10]。本研究發現,相關基因與CpG島關聯明顯,結合其他算法對相關基因啟動子重要調控元件進行預測,為下一步的研究奠定了基礎。
由于轉錄因子結合位點是一段包含在基因啟動子中的DNA序列,研究基因啟動子和轉錄因子結合位點是密不可分的。本研究選取相關基因預測的啟動子區域DNA序列進行相關基因數據分析,同樣也有很多方法和軟件/工具可供選擇。MatIspector[11]和Match是基于位置權重矩陣(PWN)來描述轉錄因子結合位點的在線工具,根據已知的轉錄因子結合位點,構建矩陣來描述轉錄因子結合位點的各個位點的堿基組成[12]。該工具可以迅速識別轉錄因子結合位點;缺點是由于背景噪音的干擾,出現許多無功能的假陽性轉錄因子結合位點[13]。Consite是基于進化發育足跡法,通過多物種間基因同源性進行交叉比對查找啟動子區保守區共同存在的轉錄因子結合部位,降低了假陽性率,使預測結果更為準確[14]。值得注意的是,轉錄因子長度較短,無論同源匹配還是模式識別,其假陽性比例都會很高[15],因此,啟動子區域識別最好基于外顯子/內顯子以及CpG島預測的結果做綜合判斷。本研究綜合基于外顯子/內顯子以及CpG島預測的結果,采用3種不同數據庫來源的在線軟件/工具并利用兩種序列分析預測方法來預測轉錄因子結合位點,可有效降低假陽性率,但仍然不能排除無實際功能的結合位點,需要進一步的實驗來驗證。
通過DNA和蛋白質序列分析預測技術,生物信息數據應用到系統進化發育預測、基因結構功能預測等方面,結果得到進一步應用和認可,同時也存在著諸多不足之處,但隨著基因組序列信息的日益豐富,計算方法和數據庫的不斷完善,可以指導實驗方向和進一步完善實驗技術,基因表達的調控機制也將逐步得到闡明。
[1]劉嵩,于皆平,劉浩,等.胃癌中PTEN基因異常甲基化的檢測[J].中華醫學雜志,2005,29(4):263-264.
[2]Bernal C,Aguayo F,Villarroel C,et al.Reprimo as a potential biomarker for early detection in gastric cancer[J].Clin Cancer Res,2008,14(19):6264-6269.
[3]李巖,李卉.DNA甲基化與胃癌相關性的研究進展[J].胃腸病學,2008,13(11):645-650.
[4]Li WQ,Pan KF,Zhang Y,et al.RUNX3 methylation and expression associated with advanced precancerous gastric lesions in a Chinese population[J].Carcinogenesis,2011,32(3):406-410.
[5]王衛政,何長華,蔡曉美,等.胃癌組織Runx3基因甲基化的檢測及意義[J].山東醫藥,2009,49(32):53-54.
[6]Takai D,Jones PA.The CpG island searcher:a new WWW resource[J].In Silico Biol,2003,3(3):235-240.
[7]Ushijima T.Detection and interpretation of altered methylation patterns in cancer cells[J].Nat Rev Cancer,2005,5(3):223-231.
[8]何克抗.計算機輔助教學研究與發展[M].北京:高等教育出版社,1996:56.
[9]Davuluri RV,Grosse I,Zhang MQ.Computational identification of promoters and first exons in the human genome[J].Nat Genet,2001,29(4):412-417.
[10]孫吉貴,韓霄松,盧欣華,等.真核生物啟動子的預測技術[J].計算機科學,2009,36(1):5-9.
[11]Cartharius K,Frech K,Grote K,et al.MatInspector and beyond:promoter analysis based on transcription factor binding sites[J].Bioinformatics,2005,21(13):2933-2942.
[12]楊科利,許強.基于堿基關聯二聯體位置權重矩陣預測酵母轉錄因子結合位點[J].生命科學研究,2008,12(02):115-120.
[13]李婷婷,蔣博,汪小我,等.轉錄因子結合位點的計算分析方法[J].生物物理學報,2008,24(5):334-347.
[14]Sandelin A,Wasserman WW,Lenhard B.ConSite:web-based prediction of regulatory elements using cross-species comparison[J].Nucleic Acids Res,2004,32(suppl 2):249-252.
[15]薛慶中.DNA和蛋白質序列數據分析工具[M].2版.北京:科學出版社,2010:45.
猜你喜歡
昆明醫科大學學報(2022年1期)2022-02-28 07:43:36
昆明醫科大學學報(2021年5期)2021-07-22 07:32:22
基層中醫藥(2020年2期)2020-07-27 02:46:06
中國組織化學與細胞化學雜志(2016年3期)2016-02-27 11:15:35
中國衛生標準管理(2015年3期)2016-01-14 03:41:46
中外醫療(2015年18期)2016-01-04 06:51:55
吉林大學學報(醫學版)(2015年4期)2015-12-17 07:48:25
醫學研究雜志(2015年6期)2015-07-01 17:40:49
醫學研究雜志(2015年9期)2015-07-01 17:28:27
中國當代醫藥(2015年20期)2015-03-01 02:04:29
