完全液相法AlOOH催化甲醇脫水行為研究
2014-08-10 12:26:51欒春暉呂經緯陰麗華
太原理工大學學報 2014年1期
關鍵詞:催化劑
欒春暉,呂經緯,陰麗華,黃 偉
(太原理工大學 a.化學化工學院; b.煤化工研究所, 太原 030024)
完全液相法AlOOH催化甲醇脫水行為研究
欒春暉a,b,呂經緯a,陰麗華b,黃 偉b
(太原理工大學 a.化學化工學院; b.煤化工研究所, 太原 030024)
用完全液相法制備了用于甲醇脫水合成二甲醚的羥基氧化鋁催化劑,對催化劑進行活性評價,并與共沉淀法、溶膠凝膠法制備的催化劑進行了對比,用XRD、NH3-TPD、XPS、氮吸附等方法對催化劑進行了表征。活性評價結果顯示,完全液相法制備的催化劑用于甲醇脫水反應,催化劑的誘導期較長,經過誘導期后甲醇轉化率可以達到80%,與傳統法相同熱處理溫度下制備的催化劑活性相當。XRD顯示,經過500℃焙燒的催化劑以γ-Al2O3形態存在;但當熱處理溫度為300℃時,無論焙燒還是液相熱處理,催化劑均以AlOOH形式存在。XPS結果表明,完全液相法制備的催化劑表面碳含量高于傳統法制備的催化劑。NH3-TPD結果表明,完全液相法制備的催化劑表面強酸中心較多,與表面碳物種有關。反應過程中甲醇和二甲醚的洗脫作用使反應后催化劑表面的碳含量有所下降,弱酸中心增加,催化活性提高。
羥基氧化鋁;催化劑;完全液相法;二甲醚;甲醇;固定床
二甲醚作為天然氣的下游產品,是重要的化工原料和有機合成中間體。由于二甲醚燃燒安全,預混氣熱值和理論燃燒溫度等性能指標優于石油液化氣,可作為城市管道煤氣的調峰氣、液化氣摻混氣,也是柴油發動機的理想燃料。二甲醚因其良好的可壓縮性可代替氟氯昂用作噴霧劑、制冷劑和發泡劑。二甲醚的制備方法主要有合成氣一步法制備工藝和甲醇脫水直接制備工藝,其中甲醇脫水法具有轉化率高、副反應少等優點[1-8]。
甲醇脫水反應采用的催化劑為固體酸催化劑。Morajkar[5]用沉淀法制備了高比表面積的γ-Al2O3,并通過吡啶紅外表征發現催化劑表面大量存在的B酸中心是催化劑具有高脫水活性的原因。Kye Sang Yoo[9]以SAPO分子篩為催化劑,對比了一系列SAPO分子篩上甲醇脫水活性的差異,發現SAPO-5和SAPO-11比SAPO-34、SAPO-18更穩定且不易積碳。V. Vishwanathan[8]研究了Na修飾的HZSM-5上粗甲醇脫水制備DME,催化劑則表現出更優異的活性,230~340 ℃范圍內甲醇轉化率達到80%,DME選擇性100%,連續生產65 h。Na的作用表現在去除表面強酸中心,減少積碳和生成烴的副反應。
甲醇脫水催化劑的制備方法有很多,常規的有沉淀法、溶膠凝膠法、微乳液法等。M. Mollavali[6]用共沉淀法合成了一系列不同n(Si)∶n(Al)的催化劑用于甲醇脫水制二甲醚,結果顯示Si改性的Al2O3具有更好的性質,比表面積隨著Si含量的增加而增加,同時表面酸性隨著n(Si)∶n(Al)比增加而增加,具有最多弱酸及中強酸中心的催化劑有最好的活性和穩定性。
完全液相法是本課題組提出的全新的催化劑制備方法[10]。在本課題組前期研究中發現,采用完全液相法制備的甲醇脫水催化劑,其脫水組分為AlOOH,而不是通常的γ-Al2O3[11]。從傳統觀點看,AlOOH一直被認為是介穩相,以AlOOH為甲醇脫水催化劑鮮見報道。對比研究表明,以完全液相法制備的AlOOH催化劑在漿態床反應器上用于甲醇脫水制備二甲醚具有較好的催化效果及優于傳統工藝的穩定性[12],而商業AlOOH用于甲醇脫水則很快失活。前期的理論研究工作表明,完全液相法制備的催化劑,由于熱處理介質不同于傳統方法的氣體氛圍,得到的催化劑在結構和成分上都與傳統法不同[13-14]。本文系在課題組相關研究基礎上,探討完全液相法制備的催化劑在固定床上的應用,以期探索完全液相法能否發展成為一種通用技術。本文采用完全液相法制備了AlOOH催化劑,研究了其在固定床反應器上催化甲醇脫水的反應情況,并與傳統方法進行了對比。
1 實驗部分
1.1 催化劑的制備
1.1.1溶膠凝膠法制備催化劑
將55 g的異丙醇鋁溶于300 mL去離子水中,加熱水解,然后加入適量濃硝酸調節pH為4~5,攪拌回流10 h至呈半透明的溶膠;將凝膠置于室溫下空氣氣氛中老化10 d,得到凝膠。凝膠置于馬弗爐中緩慢升溫焙燒,得到溶膠凝膠法制備的催化劑。焙燒溫度分別為500℃和300℃,所得催化劑分別記為A-500和A-300。
1.1.2完全液相法制備催化劑
參照溶膠凝膠法制備出溶膠,室溫下老化10 d得到凝膠。將凝膠分散于300 mL的液體石蠟中,滴入0.5 mL span80,用高剪切混合乳化機使凝膠和液體石蠟乳化,然后在40 mL/min N2保護下升溫至300℃處理10 h得漿狀催化劑。漿狀催化劑經離心分離后取固相放在索氏提取器用石油醚抽提4 d,將抽提后的催化劑置于室溫下使溶劑揮發,自然干燥后壓片造粒待用。所得催化劑記為B-300。
1.1.3沉淀法制備催化劑
稱取30 g九水硝酸鋁加入到裝有100 mL水的三口燒瓶中,再加入12.7 g無水碳酸鈉,攪拌并用NaOH溶液調節控制pH到9.沉淀結束后進行老化、抽濾、洗滌、烘干并在馬弗爐中焙燒。焙燒溫度為500℃,壓片、造粒待用。所得催化劑記為C-500。
1.2 活性評價方法
首先,將40~60目的催化劑裝入內徑為8 mm的固定床反應管中,催化劑的用量為3.0 mL。將甲醇置于鼓泡器中,保持溫度為30 ℃,經由氮氣帶入氣化室,在250 ℃預熱后進入固定床反應器進行常壓脫水。反應后進入氣液分離器,分離后的氣相采用濕式流量計計量尾氣流量,并以氣相色譜分析氣體組成。每天固定時間收集液相一次,在氣相色譜毛細管柱(固定相SE-54)上進行組分分析。
1.3 催化劑表征
樣品的XRD表征采用日本島津的XRD-6000型衍射儀,Cu Kα輻射源,Ni濾片,管電壓40 kV,電流100 mA,掃描范圍為2θ=20°~80°,掃描速度8(°)/min。NH3程序升溫脫附(NH3-TPD)采用天津市先權儀器有限公司產TP-5000多用吸附儀,催化劑裝填量為100 mg。樣品先在280℃、He氣氛下吹掃30 min,然后降溫到50 ℃脈沖吸附NH3,并吹掃物理吸附的氨氣。最后從50℃以10 ℃/min的速率程序升溫到800 ℃脫附NH3,記錄NH3脫附曲線。為防止催化劑上吸附的石蠟對氨氣的信號造成的影響,測試過程中同時用英國Hiden公司產的QIC 20質譜檢測器對尾氣進行檢測。X射線光電子能譜(XPS)分析采用VG公司的ESCALAB 250型X射線光電子能譜儀,以單色化的Al Kα為輻射源(1 486.6 eV),基礎真空7.0×10-8Pa,以Zn2p3/2(Eb=1021.8 eV)為標準校正其它元素的結合能,根據各元素的特征峰的峰面積和靈敏度因子求算催化劑表面的元素組成。比表面積和孔結構由 Tristar 3000型吸附儀測定,采用 N2為吸附質,在液氮溫度 (77 K)下進行吸附。樣品測量前在200℃抽真空8 h以上,以排除內部水分和雜質氣體。總的比表面積通過對 BET方程進行線性回歸確定,總的孔容由 BJH模型確定。
2 結果與討論
2.1 催化劑活性評價結果
對制備的4種催化劑進行活性評價,結果如圖1所示。催化劑評價結果表明,上述4個催化劑上甲醇脫水反應的二甲醚選擇性都大于99%,故這里僅討論轉化率的變化規律。轉化率結果顯示,催化劑焙燒溫度對活性有影響:500℃焙燒的沉淀法及溶膠凝膠法的催化劑甲醇轉化率都達到85%左右,其中溶膠凝膠催化劑在評價的18 d內活性基本穩定,略有下降;溶膠凝膠法300℃焙燒的催化劑比500℃焙燒的活性低,說明兩者結構上有差異。完全液相催化劑由于受熱介質石蠟沸點限制,常壓下熱處理溫度僅能達到300℃,其活性規律不同于溶膠凝膠法制備的催化劑,表現為反應初期轉化率活性很低,但隨著反應的進行,其活性逐漸提高,直到達到300℃焙燒的催化劑的活性水平后基本保持穩定,不再變化。這些現象表明催化劑在反應過程中持續發生著一些變化,并且這種變化是朝著催化劑活性提高的方向進行的,完全液相催化劑在500 h內活性持續提高,之后達到穩定,表明該工藝制備的催化劑有一個較長的誘導期,其原因應該與催化劑表面狀態有關。

反應溫度290℃,載氣流速50 mL/min
2.2 催化劑表征結果
2.2.1XRD表征
XRD對比結果表明,無論是沉淀法還是溶膠凝膠法制備的催化劑,500℃焙燒的催化劑中鋁以γ-Al2O3晶型存在,而300℃焙燒的催化劑的鋁以擬薄水鋁石形式存在,兩種形態活性上有差異。由于常壓下石蠟熱處理溫度僅能達到300℃,故完全液相工藝制備的催化劑的活性也僅能達到300℃焙燒熱處理后的水平,而無法達到更高,這是常壓液相熱處理的局限性造成的。完全液相工藝制備的催化劑反應前后的XRD圖上沒有明顯差異,說明其反應后活性的提高不是由于晶型變化引起的。同時XRD結果并未顯示出焙燒法和液相處理的催化劑晶型上有差異。

圖2 催化劑X射線衍射圖
2.2.2XPS 表征
通過X光電子能譜考察了催化劑表面元素分布(見表1),結果顯示催化劑表面都存在碳元素,其中以溶膠凝膠法和沉淀法制備的催化劑表面C元素含量較低,一般樣品在進行XPS測定時或會存在一些少量碳元素在催化劑表面,這可能是污染碳產生的信號。溶膠凝膠法制備的催化劑A-500反應后碳含量增高,可能是反應過程中催化劑表面積碳所致,表現為催化劑反應后由原來白色變為灰黑色。完全液相法制備的催化劑B-300反應后表面碳含量比反應前有所下降,在熱處理過程中有部分分解的石蠟被吸附在催化劑外表面,雖然經過石油醚的4 d抽提,表面殘存的碳仍然覆蓋了相當數量的酸中心,甚至堵塞了部分孔道,使催化劑表面利用率較低。在反應過程中反應原料甲醇以及產物二甲醚不斷地沖刷著催化劑表面,使得這些附著于催化劑表面的殘存碳物種被逐漸洗脫下來,催化劑活性也就逐漸升高,直到表面殘存碳量和焙燒法基本相當,表面殘存的碳物種基本被洗脫干凈了,催化劑活性就基本穩定了。反應前后的催化劑C 1sXPS能譜對比也反映出上述過程存在的可能性,反應前完全液相催化劑B-300表面的碳以多種形式存在,對比文獻[15-16]可知284.8 eV、288.8 eV分別對應C-C、-COOH兩種形態,第三種結合能291.8 eV對應的碳理論上應該與電負性較大的原子如Cl、F相連。但體系中沒有這些原子的來源,目前還未能確定其歸屬。沉淀法制備的C-500也有288.9 eV的峰,而反應后的B-300以及溶膠凝膠法制備的催化劑上碳主要以C-C形式存在。
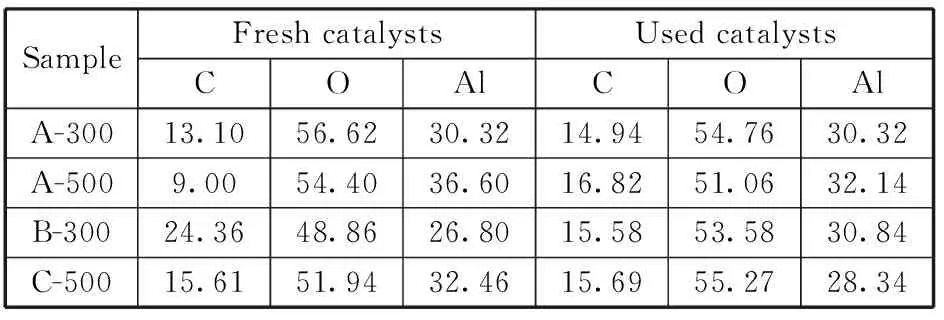
表1 催化劑表面元素質量分數 %

圖3 催化劑C1s XPS能譜對比
2.2.3NH3-TPD表征
催化劑表面酸中心的情況通過NH3-TPD考察。圖4顯示,不同的制備工藝生成的催化劑的表面酸中心情況有明顯差別。溶膠凝膠法制備的催化劑僅存在弱酸中心,但500℃焙燒的催化劑比300℃焙燒的催化劑酸中心少;完全液相法制備的催化劑B-300有弱酸中心也有強酸中心,反應后弱酸中心增多,強酸中心減少。文獻表明弱酸中心是甲醇脫水生成二甲醚的活性中心,弱酸中心增多是其活性提高的重要原因。結合XPS結果可見其強酸中心與表面存在的-COOH相關。隨著反應的進行,表面碳物種被洗脫,釋放出清潔表面,Al連接的羥基產生的弱酸中心顯露出來,因此反應后強酸中心減少,弱酸中心增多,活性提高。沉淀法制備的催化劑表面C也有兩種形態,可能與沉淀劑Na2CO3有關,相應的催化劑也有弱酸中心、中強酸中心和強酸中心。酸中心數量上與甲醇脫水活性不能簡單關聯,說明除了弱酸中心數量影響催化劑活性外,Al存在的形式是AlOOH還是Al2O3對甲醇脫水活性也有影響,甚至其影響比酸中心數量還要大。

圖4 催化劑的NH3程序升溫脫附結果
2.2.4N2吸附測定催化劑織構性質
催化劑織構性質見表2。可看出完全液相法制備的催化劑的比表面積比溶膠凝膠法低,但高于沉淀法。完全液相法催化劑反應后比反應前比表面積顯著增加,比孔容增大,平均孔徑減小,甚至明顯高于溶膠凝膠法300 ℃焙燒制備的催化劑,說明熱處理環節石蠟熱介質起到了一定的造孔劑的作用。結合表面C含量結果以及催化劑活性評價結果可以認為,完全液相催化劑在制備過程中石蠟由于毛細作用進入到催化劑的小孔中,隨著反應的進行甲醇和二甲醚對石蠟有溶解作用,帶著其離開小孔,隨著載氣脫離催化劑;長時間反應的結果,殘留在催化劑
小孔中的碳物種逐漸被帶出,留下清潔小孔,使得催化劑反應后的平均孔徑下降,比孔容增加,這是完全液相催化劑隨著反應進行活性升高的主要原因。
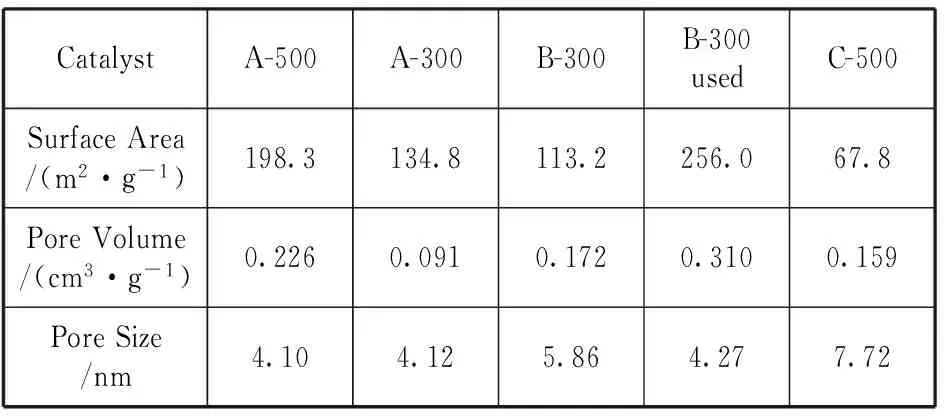
表2 甲醇脫水催化劑表面織構性質
3 結論
利用完全液相法制備了AlOOH催化劑,并用于甲醇脫水反應,經活性評價,結合多種表征手段得到以下結論:
1) 完全液相工藝制備的單組分催化劑有不同于傳統方法催化劑的結構特點和表面特征,其在固定床上甲醇脫水反應中表現出良好的穩定性。本研究認為,完全液相催化劑制備工藝可以發展成為一種具有普遍性的催化劑通用制備方法。
2) 用完全液相法制備的Al2O3催化劑用于甲醇脫水反應,經過較長的誘導期后活性基本可以達到相同熱處理溫度下傳統法催化劑的活性水平。
3) 液體石蠟熱處理的催化劑表面碳含量較高,覆蓋了活性中心,經過反應初期的甲醇及二甲醚蒸汽的多次洗脫,表面碳物種被帶離催化劑表面,活性得以提高。
4) 影響甲醇脫水活性的因素有鋁的存在形式以及表面弱酸中心數量,前者是最主要因素。
5) 完全液相法催化劑表面的碳主要以C-C形式存在,少量以-COOH形式存在,-COOH與催化劑表面強酸中心有關。
[1] 譚猗生,解紅娟,崔海濤,等. 漿態床二甲醚合成中V2O5、Sm2O3對脫水組分γ-Al2O3的修飾作用[J]. 燃料化學學報,2005, 33(5): 602-606.
[2] 張海濤,房鼎業,應衛勇,等. 甲醇氣相脫水制二甲醚本征動力學研究[J]. 天然氣化工,2009, 34(5): 9-12.
[3] 陳程雯,楊振威,林敬東,等. 高嶺土催化甲醇脫水制二甲醚的研究[J]. 廈門大學學報:自然科學版,2004, 43(5): 661-664.
[4] Yaripour F, Baghaei F, Schmidt I, et al. Catalytic dehydration of methanol to dimethyl ether (DME) over solid-acid catalysts[J]. Catalysis Communications,2005, 6(6): 147-152.
[5] Morajkar P P, Fernandes J B. A new facile method to synthesize mesoporous γ-Al2O3of high surface area and catalytic activity [J]. Catalysis Communications,2010, 11: 414-418.
[6] Mollavali M, Yaripour F, Mohammadi-Jam S, et al. Relationship between surface acidity and activity of solid-acid catalysts in vapour phase dehydration of methanol.[J]. Fuel Processing Technology,2009, 90(9): 1093-1098.
[7] Topp-Jorgensen J. Catalysts for Use in Ether Synthesis:Europe, EP1984-308989[P]. 1985-07-17.
[8] Vishwanathan V, Jun K, Kim J, et al. Vapor phase dehydration of crude methanol to dimethyl ether over Na-modified HZSM-5 catalysts[J]. Applied Catalysis Α: General,2004, 276(1/2): 251-255.
[9] Yoo K S, Kim J, Park M, et al. Influence of solid acid catalyst on DME production directly from synthesis gas over the admixed catalyst of Cu/ZnO/Al2O3and various SAPO catalysts[J]. Appl Catal:A,2007, 330: 57-62.
[10] 黃偉,高志華,陰麗華,等. 一種漿狀催化劑及其制備方法:中國,200610102268.1[Ρ].2007-06-06.
[11] Gao Z, Huang W, Yin L, et al. Liquid-phase preparation of catalysts used in slurry reactors to synthesize dimethyl[J]. Fuel Processing Technology,2009, 90: 1442-1446.
[12] Liu L, Huang W, Gao Z, et al. The dehydration of methanol to dimethyl ether over a novel slurry catalyst.[J]. Energy Sources:Part A, 2010, 32(15): 1379-1387.
[13] Zuo Z, Huang W, Han P, et al. Solvent effects for CO and H2adsorption on Cu2O (111) surface A:density functional theory study[J]. Applied Surface Science,2010, 256: 2357-2362.
[14] Zuo Z, Sun L, Huang W, et al. Surface properties of copper in different solvent mother solutions:A density functional theory study[J]. Applied Catalysis A: General,2010, 375: 181-187.
[15] 張轉芳,閆爾云. 純化條件對多壁碳納米管功能化的影響[J]. 電子元件與材料, 2013, 32(4): 32-35.
[16] Ying Y, Li-Li M, Wen-Ya H, et al. Sonication assisted deposition of Cu2O nanoparticles on multiwall carbon nanotubes with polyol process[J]. Carbon,2005, 43: 670-673.
(編輯:張紅霞)
CatalysisPerformanceofAlOOHCatalystsPreparedbyCompleteLiquid-PhaseMethodforMethanolDehydrationtoDimethylEtherinaFixedBed
LUANChunhuia,b,LVJingweia,YINLihuab,HUANGWeib
(a.CollegeofChemistryandChemicalEngineering;b.InstituteofCoalChemistryEngineering,TaiyuanUniverisityofTechnology,Taiyuan030024,China)
In this work, the AlOOH catalysts prepared by complete liquid-phase technology was investigated in a fixed bed reactor for methanol dehydration and compared with the catalysts prepared by traditional method, such as precipitation and sol gel method. These catalysts were characterized by XRD, NH3-TPD, XPS and N2adsorption. Results show that the convertion of methanol was 80%, after longer induction period. XRD characterization reveals that the Al element existed in the form of γ-Al2O3after calcination at 500 ℃,but when the heat treatment temperature was 300 ℃, the catalysts existed in the form of AlOOH. More carbon covered the complete liquid-phase catalyst,as proved by XPS. NH3-TPD results show that the more strong acid sites were associated with the surface carbon. Elution of methanol and DME in the reaction process removed the surface carbon on the catalyst surface, increased the number of weak acid sites and improved catalytic activity in the fixed reactor.
AlOOH;catalyst;complete liquid-phase;DME;methanol;fixed bed reactor
2013-08-06
國家自然科學基金資助項目(21176167)
欒春暉(1969-),女, 江蘇泰州人,博士,副教授,主要從事催化方面研究,(Tel)13073529816
黃偉,博士,教授,博導,(Tel)0351-6018703
1007-9432(2014)01-0092-05
O643
:A
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
