La摻雜對Cr2O3催化劑氣相氟化四氯乙烯反應(yīng)性能的影響
2014-10-13 07:57:52范鏡蓮程永香趙洋王芳羅孟飛王月娟
化工進(jìn)展 2014年1期
關(guān)鍵詞:催化劑
范鏡蓮,程永香,趙洋,王芳,羅孟飛,王月娟
(浙江師范大學(xué)物理化學(xué)研究所,浙江省固體表面反應(yīng)化學(xué)重點(diǎn)實(shí)驗(yàn)室,浙江 金華 321004)
五氟乙烷(簡稱HFC-125)的消耗臭氧潛能值(ODP)為0,全球變暖潛能值(GWP)為2800,是R404A、R407C、R410、R402A、R507等環(huán)保型混合制冷劑的主要組分,這些環(huán)保型混合制冷劑可
以替代現(xiàn)有的二氟一氯甲烷(HCFC-22)制冷劑。HFC-125還廣泛用作發(fā)泡劑、溶劑、噴射劑和干蝕刻劑[1]。
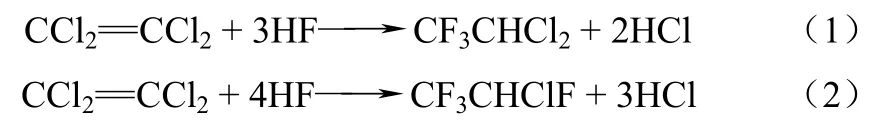
氟化含氯烯烴是合成氫氟烴(HFCs)的主要合成路線,如以三氯乙烯為原料合成 HFC-134a(CF3CH2F)[2];四氯乙烯為原料合成HFC-125[3-4]。工業(yè)上通常采用液相和氣相氟化兩種氟化路線,前者由于對設(shè)備腐蝕性大、環(huán)境污染嚴(yán)重逐漸被淘汰[4],因此目前普遍采用的是氣相氟化路線。四氯乙烯路線分兩個步驟來實(shí)現(xiàn):第一步是氟化四氯乙烯(簡稱PCE)制備HCFC-123(2,2-二氯-1,1,1-三氟乙烷)和HCFC-124(2-氯-1,1,1,2-四氟乙烷),反應(yīng)式如式(1)、式(2)。
第二步由HCFC-123或HCFC-124進(jìn)一步氟化制備HFC-125,反應(yīng)如式(3)、式(4)。
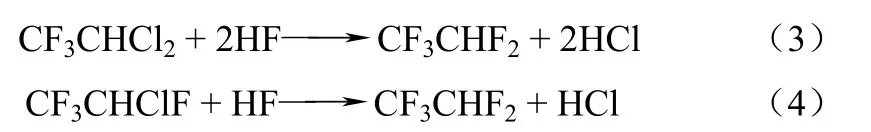
氣相氟化普遍采用 Cr基催化劑,有負(fù)載型Cr/AlF3[4]、Cr/Al2O3[5]、Cr/MgO 和 Cr/MgF2[6]等催化劑;非負(fù)載的CrOx[7]催化劑;摻雜的Cr2O3[8]催化劑。雖然這些催化劑已經(jīng)廣泛用于HFCs的工業(yè)化合成,但是對催化劑的活性物種及其反應(yīng)機(jī)理尚沒有統(tǒng)一的認(rèn)識。很多研究認(rèn)為催化劑表面CrOF是氣相氟化反應(yīng)的活性中心,如Lee等[6]研究發(fā)現(xiàn),Cr/MgF2催化劑活性要比純CrOx催化劑活性高,他們認(rèn)為催化劑的活性中心為 CrOxFy,而非 CrF3。Adamczyk等[9]用(NH4)3CrF6和Cr2O3在高溫下制備了一系列的CrOxFy催化劑,并用XRD和XANES對催化劑進(jìn)行了表征,實(shí)驗(yàn)表明含有CrOxFy物種催化劑表現(xiàn)出比純Cr2O3和CrF3更高的活性,是該催化劑的活性中心。Rao等[10]制備了 Cr2O3、Cr2O3/Al2O3(浸漬法)及 Cr2O3/Al2O3(共沉淀法)催化劑,用 XPS技術(shù)對 Cr物種進(jìn)行了表征,認(rèn)為Cr(OH)F2為活性物種。另外,有人認(rèn)為催化劑中高價態(tài)的 Cr有利于氟化反應(yīng)[11],還有人認(rèn)為催化劑的活性依賴于催化劑表面的酸性位[12-13]。總而言之,目前關(guān)于 Cr基催化劑的活性物種還沒有一致的認(rèn)識,還需要進(jìn)一步的考察和研究。
先前作者[14]報(bào)道了ZnO/Al2O3催化劑對氣相氟化四氯丙烯反應(yīng)性能的催化性能,考察了載體Al2O3的焙燒溫度對催化劑性能的影響,發(fā)現(xiàn)Al2O3載體經(jīng)過1100 ℃焙燒制得的ZnF2/Al2O3催化劑催化性能最高,當(dāng)反應(yīng)溫度為300 ℃時,四氯乙烯的轉(zhuǎn)化率為 45.7%,HCFC-123(2,2-二氯-1,1,1-三氟乙烷)和HCFC-124(2-氯-1,1,1,2-四氟乙烷)的總選擇性為 48.2%。然而,催化劑的轉(zhuǎn)化率和選擇性較低,無法滿足工業(yè)化需要。
本文以 Cr2O3為基礎(chǔ),通過向其中添加不同含量的La助劑,研究不同含量的La助劑摻雜對Cr2O3催化劑氟化四氯乙烯性能的影響及其表面酸性、CrOxFy物種對催化劑活性的影響。
1 實(shí)驗(yàn)部分
1.1 實(shí)驗(yàn)試劑
九水硝酸鉻,AR,國藥集團(tuán)化學(xué)試劑有限公司;六水硝酸鑭,AR,國藥集團(tuán)化學(xué)試劑有限公司;濃氨水,AR,杭州余杭利人醫(yī)藥化工有限公司;四氯乙烯,純度>99%,浙江三美化工股份有限公司提供;無水HF,純度>99.9%,浙江三美化工股份有限公司;蒸餾水,自制。
1.2 催化劑制備
La2O3-Cr2O3催化劑采用沉淀法制備。稱取一定量的 Cr(NO3)3·9H2O 和 La(NO3)3·6H2O 配成混合溶液,然后用沉淀劑氨水沉淀,分離得到的沉淀物經(jīng)烘干(120 ℃),在500 ℃氮?dú)鈿夥障卤簾? h,即制得xLa2O3-Cr2O3催化劑前體,x為催化劑中La摩爾分?jǐn)?shù),分別為0、1%、5%和10%。將La2O3-Cr2O3催化劑前體裝入自制的反應(yīng)裝置(內(nèi)徑為10 mm的不銹鋼反應(yīng)管),通入N2-HF混合氣體(HF與 N2的摩爾比為4,總流量為50 mL/min),升溫至260 ℃保持1 h,再升溫至400 ℃保持3 h,得到氟化后的催化劑,催化劑記為 xLaF3-Cr2O3。Cr2O3(F)、1LaF3-Cr2O3、5LaF3-Cr2O3、10LaF3-Cr2O3催化劑的比表面積分別為53 m2/g、49 m2/g、47 m2/g和42 m2/g。
LaF3采用LaF3-Cr2O3催化劑相同的方法制備。氟化預(yù)處理后的樣品記為LaF3,LaF3的比表面積為12 m2/ g。
CrF3的制備:稱取一定量的Cr(NO3)3·9H2O配成溶液,然后用沉淀劑氨水沉淀,分離得到的沉淀物經(jīng)烘干(120 ℃),即制得 Cr(OH)3前體。將Cr(OH)3前體裝入自制的反應(yīng)裝置(內(nèi)徑為10 mm的不銹鋼反應(yīng)管),經(jīng)200 ℃干燥2 h后,在N2-HF混合氣體(HF與 N2的摩爾比為 4,總流量為 50 mL/min)下升溫至 300 ℃氟化 10 h得到無定形CrF3,其比表面積為68 m2/g。
1.3 催化劑表征
X射線衍射(XRD)分析采用荷蘭Philips公司生產(chǎn)的PW3040/60型全自動X射線衍射儀。Cu Kα射線,管電壓40 kV,管電流40 mA,掃描速度0.15°/s,掃描范圍2θ 為10°~80°。所有XRD測試均在靜態(tài)空氣氣氛下進(jìn)行。
催化劑的比表面積測定采用美國Quantachrome Autosorb-1型N2物理吸附儀測定。所有測試樣品在300℃真空預(yù)處理4h,與液氮溫度下進(jìn)行實(shí)驗(yàn)。采用BET公式計(jì)算樣品的比表面積。
催化劑的表面酸性通過氨氣程序升溫脫附(NH3-TPD)實(shí)驗(yàn)來測試。實(shí)驗(yàn)在自制的裝置上進(jìn)行,將100 mg催化劑裝入直徑為6 mm的石英管反應(yīng)器中,于350 ℃氮?dú)鈿夥障赂稍?.5 h,除去催化劑中的水分,然后降溫至50 ℃吸附NH330 min,再于100 ℃下氮?dú)獯祾?0 min以除去殘留的和物理吸附的 NH3。然后以 10 ℃/min的速率升溫至600 ℃進(jìn)行NH3脫附,脫附的NH3通過TCD檢測器檢測。
X射線光電子能譜是在VGESCALAB MK-2型能譜儀上測定,AlKα射線源(1486.6 eV),加速電壓20 kV,樣品以C1s=284.6 eV為基準(zhǔn)進(jìn)行結(jié)合能校正。
1.4 催化劑活性評價
在自制的內(nèi)徑為 10 mm的不銹鋼管固定床反應(yīng)器中裝入3 mL催化劑,熱電偶放在催化劑床層的中間監(jiān)控反應(yīng)溫度。控制 HF氣體的流量為 20 mL/min,反應(yīng)物中HF與四氯乙烯摩爾比為13∶1。反應(yīng)物和產(chǎn)物用氣相色譜(島津GC-2014)進(jìn)行在線分析,使用氫離子火焰檢測器(FID)和GS-GASPRO毛細(xì)管柱(60 m×0.32 mm)。
2 結(jié)果與討論
2.1 催化劑的物相分析
圖 1為 La2O3-Cr2O3和 LaF3-Cr2O3催化劑的XRD圖譜。對于La2O3-Cr2O3催化劑[圖1(a)],Cr2O3和1La2O3-Cr2O3樣品只觀察到Cr2O3的特征衍射峰,表明Cr物種以Cr2O3的形式存在,沒有檢查到La的相關(guān)衍射峰,表明低含量的 La以無定形的形式存在,或含量太低XRD檢測不到。當(dāng)La含量達(dá)到5%時,除了Cr2O3的特征衍射峰,還觀察到LaCrO4和LaCrO3的特征衍射峰,表明La含量的增加,有利于形成La-Cr復(fù)合氧化物。從La2O3-Cr2O3催化劑前體經(jīng)氟化預(yù)處理后的 LaF3-Cr2O3催化劑[圖 1(b)]的XRD圖可以看出,催化劑中Cr2O3的特征衍射峰依然存在,這說明在氟化預(yù)處理的過程中晶相Cr2O3很難被氟化成 CrF3,這與文獻(xiàn)中報(bào)道是一致的[10,15]。文獻(xiàn)[16]中指出,由 Cr2O3和 HF反應(yīng)制CrF3,該反應(yīng)具有較小負(fù)值的吉普斯自由能(ΔrG=?68 kJ/mol)。這同樣也說明該反應(yīng)也較難進(jìn)行或者說該反應(yīng)需要較長的時間才能有較大程度的反應(yīng)。預(yù)氟化后的催化劑除了有Cr2O3特征衍射峰外,同時還出現(xiàn)了LaF3的特征衍射峰,并且這些峰隨著 La含量的增加變得越來越強(qiáng),然而,LaCrO4和LaCrO3的特征衍射峰卻消失了,表明LaCrO4和LaCrO3完全轉(zhuǎn)化成LaF3和CrF3(或Cr2O3)。由于La2O3容易氟化生成LaF3[17],因此1LaF3-Cr2O3催化劑中沒有觀察到 LaF3的特征衍射峰可能與助劑的添加量較少,生成的 LaF3均勻分散在 Cr2O3表面有關(guān)。
圖2是LaF3-Cr2O3催化劑的Raman圖譜。對于LaF3-Cr2O3催化劑,從圖2中可以看出,在302 cm?1、350 cm?1、550 cm?1和 611 cm?1處出現(xiàn) 4 個 Raman振動峰,這4個峰都?xì)w屬為晶相Cr2O3的Raman振動峰[18]。這說明通過氟化預(yù)處理后Cr2O3依然存在,這與XRD結(jié)果是一致的,進(jìn)一步說明晶相的Cr2O3很難被氟化。

圖1 La2O3-Cr2O3和LaF3-Cr2O3催化劑的XRD圖譜
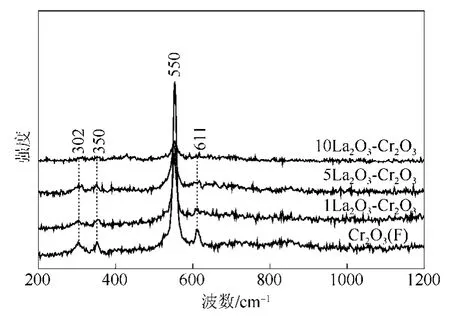
圖2 LaF3-Cr2O3催化劑的Raman圖譜
2.2 催化劑的NH3-TPD表征
圖 3是 Cr2O3(F)和 LaF3-Cr2O3催化劑的 NH3-TPD圖,為了對比,圖3中也給出了LaF3和CrF3的NH3-TPD圖。從圖3中可以看出,Cr2O3(F)催化劑在150~550 ℃之間出現(xiàn)分布范圍較寬的NH3脫附峰,表明酸性位分布較廣,包含弱酸性位和強(qiáng)酸性位。1LaF3-Cr2O3催化劑的NH3脫附峰與Cr2O3(F)催化劑基本相同。仔細(xì)觀察可以發(fā)現(xiàn),1LaF3-Cr2O3催化劑低于200 ℃沒有發(fā)現(xiàn)NH3脫附,表明部分弱酸中心消失。然而,當(dāng)La添加量增加到5%和10%時,在450 ℃附近出現(xiàn)一個很強(qiáng)的NH3脫附峰,低溫區(qū)(170~300 ℃)的 NH3脫附峰消失,催化劑表面酸性位分布變窄,峰面積明顯增加。這表明隨著 La含量的增加,LaF3-Cr2O3催化劑弱酸中心減少,催化劑表面酸性位的性質(zhì)發(fā)生改變,催化劑表面只有強(qiáng)酸中心。通過對 NH3-TPD過程流出NH3的定量計(jì)算,Cr2O3(F)、1LaF3-Cr2O3、5LaF3-Cr2O3、10LaF3-Cr2O3、LaF3和CrF3樣品表面脫附的NH3(表面酸量)分別為 83 μmol/g、58 μmol/g、100 μmol/g、232 μmol/g、78 μmol/g 和 346 μmol/g。根據(jù)催化劑的表面積和表面酸量可計(jì)算得到催化劑的表面酸密度。Cr2O3(F)、1LaF3-Cr2O3、5La F3-Cr2O3和10La F3-Cr2O3的表面酸密度分別為1.57 μmol/m2、1.18 μmol/m2、2.13 μmol/m2和 5.52 μmol/m2;LaF3和 CrF3的表面酸密度分別為 6.50 μmol/m2和 5.09μmol/m2。可見少量La的添加使得表面酸密度減少;La添加量達(dá)到5%后,由于生成較多的LaF3和CrF3,催化劑表面酸量明顯增加。
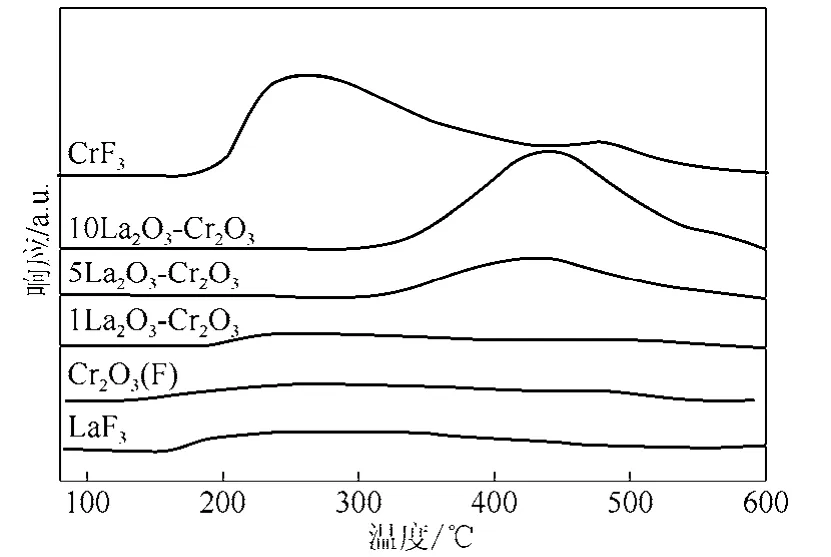
圖3 Cr2O3(F)和LaF3-Cr2O3催化劑的NH3-TPD圖
2.3 催化劑的XPS表征
圖4是LaF3-Cr2O3催化劑的Cr2p XPS圖譜。由圖4可以看到,在結(jié)合能為575.5 eV和577.5 eV分別出現(xiàn)了兩個峰值,這些峰值的出現(xiàn)歸功于Cr3+的2p3/2電子,根據(jù)文獻(xiàn)[11,19],峰值為575.5 eV和577.5 eV的兩個峰分別歸屬為Cr2O3和CrOxFy,可見LaF3-Cr2O3催化劑表面存在Cr2O3和CrOxFy。表 1是 LaF3-Cr2O3催化劑表面 CrOxFy相對含量和Cr2p2/3的結(jié)合能。催化劑表面CrOxFy含量由圖4中CrOxFy的峰面積與Cr2O3和CrOxFy兩個峰的總面積相比計(jì)算得到。從表1中可以看出,隨著LaF3-Cr2O3催化劑中 La含量的增加,CrOxFy物種的結(jié)合能往更高結(jié)合能方向偏移,這可能是因?yàn)?La的含量與Cr2O3的相互作用造成的。1LaF3-Cr2O3催化劑的表面 CrOxFy含量(35.7%)低于 Cr2O3(F)催化劑表面CrOxFy含量(36.5%),然而進(jìn)一步增加La的含量,催化劑表面CrOxFy含量增加,這表明La的添加量影響LaF3-Cr2O3表面CrOxFy的含量。
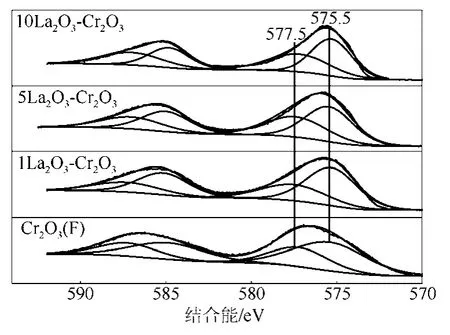
圖4 LaF3-Cr2O3催化劑的Cr2p XPS圖譜
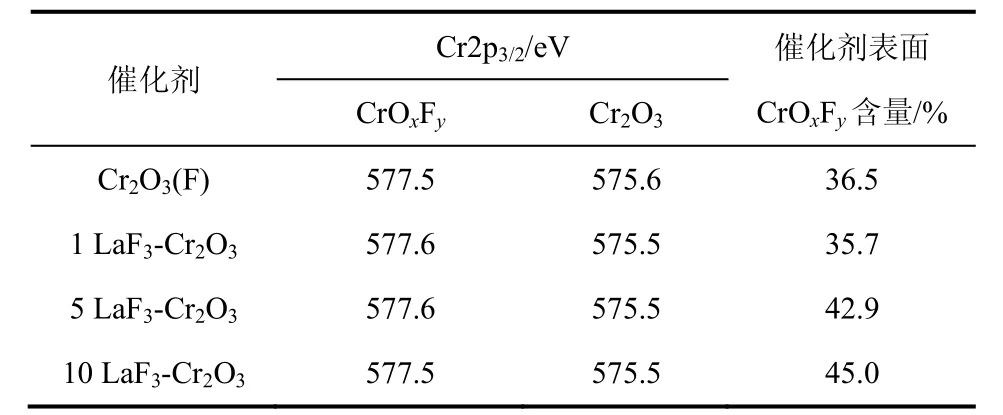
表1 LaF3-Cr2O3催化劑表面CrOxFy含量和Cr 2p2/3的結(jié)合能
2.4 LaF3-Cr2O3催化劑催化劑的四氯乙烯氟化反應(yīng)性能
表2列出了LaF3-Cr2O3催化劑的四氯乙烯氟化反應(yīng)的轉(zhuǎn)化率、選擇性和反應(yīng)比速率(數(shù)據(jù)取自反應(yīng)10 h)。從表2中可以看出,隨著La含量的增加,PCE的轉(zhuǎn)化率逐漸增加,由Cr2O3催化劑的81.1%到10LaF3-Cr2O3催化劑的98.7%,催化劑的比速率總體上隨著催化劑表面酸密度的增加而增加。然而,HCFC-123、HCFC-124和HFC-125的總選擇性卻先增加后減少。其中1LaF3-Cr2O3催化劑上HCFC-123、HCFC-124和HFC-125的總選擇性最高(90.1%)。然而隨著 La含量的繼續(xù)增加,HCFC-123、HCFC-124和HFC-125的總選擇性快速下降,當(dāng)La含量為10%(摩爾分?jǐn)?shù))時,10LaF3-Cr2O3催化劑上HCFC-123、HCFC-124和HFC-125的總選擇性僅為 34.7%。同時,隨著 La含量的增加,副產(chǎn)物HCFC-133a的選擇性逐漸增加,當(dāng)La含量為10%(摩爾分?jǐn)?shù))時,HCFC-133a的選擇性高達(dá)31.4%。
將催化劑表面酸密度與HCFC-123、HCFC-124和HFC-125選擇性總和作圖,得到催化劑表面酸量與產(chǎn)物總選擇的關(guān)系圖(圖5)。從圖5中可以看出,隨著酸量的增加,HCFC-123 、HCFC-124和HFC-125的總選擇性逐漸減少。這說明催化劑表面酸中心的增加不利于 HCFC-123、HCFC-124和HFC-125產(chǎn)物的生成。從圖5還可以看出,1LaF3-Cr2O3催化劑表面酸密度低于 Cr2O3(F),有利于HCFC-123、HCFC-124和 HFC-125的生成;當(dāng)La添加量達(dá)到 5%后,催化劑表面酸量明顯增加,提高了反應(yīng)比速率,但不利于HCFC-123、 HCFC-124和 HFC-125的生成。也發(fā)現(xiàn) HCFC-133a和 HFC-134a等副產(chǎn)物隨著催化劑酸量的增加而增加,其中HCFC-133a的量增加尤其明顯,當(dāng)催化劑添加了5%的La時,HCFC-133a的選擇性為16.4%,當(dāng)催化劑添加了 10%的 La時,HCFC-133a的選擇性為31.4%,這說明表面酸密度與目標(biāo)產(chǎn)物(HCFC-123、HCFC-124和 HFC-125)總選擇性存在對應(yīng)關(guān)系,即催化劑表面相對酸密度的增加不利于HCFC-123、HCFC-124和HFC-125的生成。

表2 LaF3-Cr2O3上PCE的轉(zhuǎn)化率,HCFC-123、HCFC- 124、HFC-125的選擇性和反應(yīng)的比速率(數(shù)據(jù)取自反應(yīng)10 h)
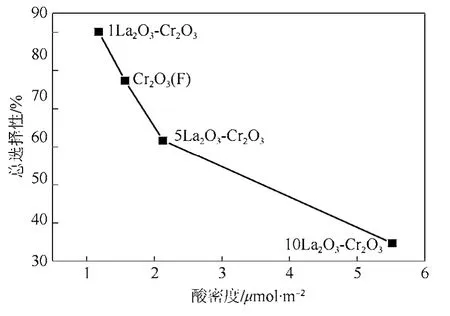
圖5 催化劑表面酸密度與產(chǎn)物總選擇(HCFC-123 +HCFC-124+ HFC-125)的關(guān)系圖
催化劑表面酸性增加往往有利于歧化反應(yīng)的發(fā)生[13,20-21],Hess 等[12]通過比較 β-AlF3和 AlF2(OH)催化劑的 CHClF2歧化反應(yīng),發(fā)現(xiàn)表面酸中心有利于歧化反應(yīng)的發(fā)生。然而表面酸性也有利于裂解反應(yīng)的發(fā)生[22-25]裂解反應(yīng)不僅需要酸性活性中心,而且能斷裂C—C鍵的強(qiáng)酸中心[26]。由于催化劑表面酸性增加有利于歧化和裂解反應(yīng),因此過多的 La使得催化劑表面酸性增加從而導(dǎo)致副反應(yīng)的發(fā)生,使得有效產(chǎn)物(HCFC-123、HCFC-124和HFC-125)的選擇性下降。
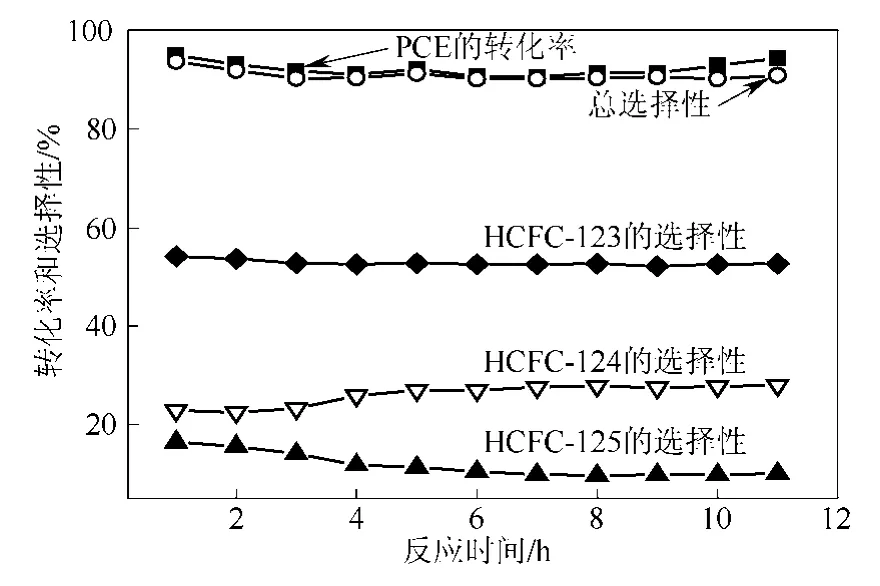
圖6 1LaF3-Cr2O3催化劑上氟化PCE的反應(yīng)穩(wěn)定性(反應(yīng)溫度為300 ℃)
圖6是在300 ℃下1LaF3-Cr2O3催化劑上氟化PCE反應(yīng)的穩(wěn)定性測試結(jié)果。從圖6中可以看出,PCE的轉(zhuǎn)化率和HCFC-123、HCFC-124和HFC-125的總選擇性在反應(yīng)開始前3 h有輕微的下降,PCE的轉(zhuǎn)化率由 94.9%降到 91.0 %,HCFC-123、HCFC-124和 HFC-125的總選擇性由 93.6%降到90.1%。在此之后,PCE和HCFC-123、HCFC-124和HFC-125的總選擇性保持穩(wěn)定,這說明該催化劑具有良好的穩(wěn)定性。
3 結(jié) 論
LaF3-Cr2O3催化劑具有較好的催化氟化四氯乙烯反應(yīng)性能,隨著La摻雜含量的增加,LaF3-Cr2O3催化劑上 PCE的轉(zhuǎn)化率不斷升高,而有效產(chǎn)物(HCFC-123 + HCFC-124 + HFC-125)總選擇性先升后降。當(dāng)La摻雜為1%時,1LaF3-Cr2O3催化劑性能最佳,PCE的轉(zhuǎn)化率和有效產(chǎn)物總選擇性分別為92.8%和90.1%。結(jié)果表明,催化劑表面強(qiáng)酸中心不利于有效產(chǎn)物的生成。
[1]劉坤峰,楊會娥,張文慶,等. 氣相法合成五氟乙烷(HFC-125)的研究進(jìn)展[J]. 有機(jī)氟工業(yè),2009(1):55-58.
[2]Quan H D,Li Z,Zhao Z X,et al. Preparation of 1,1,1,2-tetrafluoroethane (HFC-134a) by vapor-phase catalytic fluorination[J]. Appl. Catal. B,1996,8:209-215.
[3]Yoshimura T,Homoto Y,Yamada Y,et al. Process for producing pentafluoroethane and tetrafluoroethane:US,6011185[P].2000-01-04.
[4]Cheminal B,Lacroix E,Lants A,et al. Process for fluorination of perchloroethylene or of pertachloroethane:US,5932776[P].1999-08-03.
[5]Cho D H,Kim Y G,Chung M J,et al. Preparation and characterization of magnesia-supported chromium catalysts for the fuorination of 1,1,1-triuoro-2-chloroethane (HCFC-133a)[J]. Appl.Catal. B,1998,18:251-261.
[6]Lee H,Jeong H D,Chung Y S,et al. Fluorination of CF3CH2Cl over Cr-Mg fluoride catalyst:The effect of temperature on the catalyst deactivation[J]. J. Catal.,1997,169:307-316.
[7]Brunet S,Requieme B,Colnay E,et al. Catalytic gas-phase fluorination of 1,1,l-trifluoro-2-chloroethane over chromium(Ⅲ)oxide:Preparation of hydrofluoroalkanes[J]. Appl. Catal. B,1995,5:305-317.
[8]Krishna Murthy J,Gross U,Rüdiger S,et al. Synthesis and characterization of chromium(Ⅲ)-doped magnesium fluoride catalysts[J]. Appl. Catal. A,2005,282:85-91.
[9]Adamczyk B,Boese O,Weiher N,et al. Fluorine modified chromium oxide and its impact on heterogeneously catalyzed fluorination reactions[J]. J. Fluorine Chem.,2000,101:239-246.
[10]Rao J M,Sivaprasad A,Narsaiah B,et al. A comparative study of bulk and supported chromia catalysts for the fluorination of trichloroethylene[J]. J. Catal.,1999,184:105-111.
[11]Quan H D,Tamura M,Matsukawa Y,et al. Investiganiton into chromia-based catalyst and its application in preparing difluoromethane[J]. J. Mol. Catal. A:Chem.,2004,219:79-85.
[12]Hess A,Kemnitz E. Characterization of catalytically active sites on aluminum oxides,hydroxyfluorides,and fluorides in correlation with their catalytic Behavior[J]. J. Catal.,1994,149(2):449-457.
[13]Brunet S,Boussand B,Barrault J,et al. Catalytic fluorination over chromium oxides. Preparation of hydrofluorocarbons[J]. Stud. Surf.Sci. Catal.,1996,101:379-385.
[14]程永香,謝遵運(yùn),羅孟飛,等. 氣相氟化四氯丙烯的 ZnF2/Al2O3催化劑研究[J]. 化工進(jìn)展,2012,31(11):2483-2487.
[15]Xing L Q,Bi Q Y,Meng F L,et al. In situ Raman spectroscopy studies on chromium oxide catalyst in an anhydrous hydrogen fluoride atmosphere[J]. J. Raman Spectrosc.,2011,42:1095-1099.
[16]Bonniface D W,Scott J D,Watson M J,et al. Halogen exchange reactions for CFC alternatives:The behaviour of fluorine-18 labeled hydrogen fluoride towards prefluorianted chromia containing nickl(Ⅱ) orzinc (Ⅱ)[J]. Green Chem.,1999,1(1):9-11.
[17]Bi Q Y,Lu J Q,Meng F L,et al. Effect of calcination temperature on La-modified Al2O3catalysts for vapor phase hydrofluorination of acetylene to vinyl fluoride[J]. Chin. J. Chem. Phys.,2010,23:89-94.
[18]Barshilia H C,Rajam K S. Growth and characterization of chromium oxide coating prepared by pulsed-direct current reactive unbalanced magnetron sputtering[J]. Appl. Surf. Sci.,2008,255:2925-2931.
[19]Loustaunau A,F(xiàn)ayolle-Romelaer R,Celerier S,et al. Catalytic fluorination of various chlorinated hydrocarbons by HF and a chromium based catalyst:Effect of the presence of zinc[J]. Catal.Lett.,2010,138:215-223.
[20]Bell T N,Kirszensztejn P,Czajka B. Catalytic conversion of CC12F2on a γ-A12O3catalyst[J]. Catal. Lett.,1995,30:305-312.
[21]Hess A,Kemnitz E. Surface acidity and catalytic behavior of modified zirconium and titanium dioxides[J]. Appl. Catal. A:General,1997,149:373-389.
[22]Yusaku T,Tatsumi I. Catalytic decomposition of CFCs[J]. Catalysis Surveys from Japan,1998,2:165-173.
[23]Yusaku T,Maiko N,Rie M,et al. Decomposition of chloro ?uorocarbons over metal phosphate catalysts Part Ⅰ. Decomposition of CCl2F2over metal phosphate catalysts[J]. Phys. Chem. Chem.Phys.,1999,1:2367-2372.
[24]Masahiro T,Miki N,Yasushi F,et al. Decomposition of chlorofluorocarbons in the presence of water over zeolite catalyst[J].Appl. Catal. B:Environmental,1996,9:167-177.
[25]潘思忠,楊雪敏,高滋. 相同硅鋁比的ZSM-5、ZSM-35和Md沸石的表面酸性與催化性能[J]. 催化學(xué)報(bào),1988,9(2):158-164.
[26]Corna A. Application of zeolites in fluid catalytic cracking and related processes[J]. Stud. Surf. Sci. Catal.,1989,49:49-67.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學(xué)學(xué)報(bào)(工學(xué)版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(xué)(2015年4期)2016-01-17 09:01:27
應(yīng)用化工(2014年3期)2014-08-16 13:23:50
