肝豆狀核變性的非典型頭顱MRI表現:附2例報告及文獻復習
2015-03-11 08:26:21喻哲明鄭小敏馬昱金莉蓉
中國臨床醫學 2015年1期
關鍵詞:磁共振成像
喻哲明 鄭小敏 馬昱 金莉蓉
(復旦大學附屬中山醫院神經內科,上海 200032)
?
·論著·
肝豆狀核變性的非典型頭顱MRI表現:附2例報告及文獻復習
喻哲明鄭小敏馬昱金莉蓉
(復旦大學附屬中山醫院神經內科,上海200032)
摘要目的:探討肝豆狀核變性患者的非典型頭顱MRI表現特點。方法: 本文分析了2例經臨床生化及基因診斷的肝豆狀核變性患者的頭顱MRI表現及臨床特點,并結合文獻就已報道病例做回顧分析。結果:肝豆狀核變性患者的典型頭顱MRI表現為雙側豆狀核、丘腦、中腦等部位損害。本文中2例患者頭顱MRI均提示皮質下白質低T1WI、高T2WI及高Flair異常信號,較為少見。結合既往文獻報道,共13例肝豆狀核變性患者,頭顱MRI均提示皮質下白質異常信號,其中9例患者病灶累及大腦皮層(額葉9例、頂葉4例、顳葉2例)。13例患者中,11例有運動遲緩、活動障礙等錐體外系表現,6例智力減退或癡呆,9例有癲癇發作。此類患者癲癇發作比例較高可能與病灶累及皮質及皮質下白質相關,暫未提示特異的ATP7B基因突變位點。結論:肝豆狀核變性患者的頭顱MRI表現多變,且和臨床表現密切相關。
關鍵詞肝豆狀核變性;磁共振成像;基因
肝豆狀核變性(hepatolenticular degeneration,HLD)是一種少見的常染色體隱性遺傳病,由Wilson[1]于1921年全面描述,故又稱Wilson病(Wlison’s disease, WD)。WD患者因體內異常銅代謝而出現多種臨床表現,診斷困難。WD患者頭顱MRI異常表現亦多樣,主要表現為基底節、腦干、丘腦等部位對稱性低T1、高T2信號,可伴有異常信號強化、腦萎縮等。本研究分析了在我院經臨床生化檢查、基因檢測確診為WD的2例患者的非典型頭顱MRI表現,并進行相關文獻復習,以進一步了解該病的MRI特點。
1病例資料
病例1,男性,17歲。以“手抖、持物不穩1年”于2012年7月11日入院。患者于1年前發現雙手不自主抖動(靜止性細小震顫),持筷不穩,毛巾擰之不干,步行欲快不能,跑步困難,言語不清,飲水易嗆咳,癥狀呈進行性加重。既往史及家族史無特殊。神經系統查體:神志清楚,時間、地點、人物定向可,言語略不清;K-F環(+);四肢肌張力可,肌力Ⅴ級,腱反射(++),病理征(-),雙上肢輪替動作較差。生化檢查:外周血紅細胞、白細胞降低,血谷氨酸氨基轉移酶(ALT)、天冬氨酸轉氨基轉移酶(AST)在正常范圍;血清銅7.6 μmol/L,血銅藍蛋白<0.1 g/L,24 h尿銅3.3 μmol。基因檢測:ATP7B基因第13號外顯子Ala1003Ala雜合同義突變,見圖1A。腹部超聲未提示肝脾腫大。頭顱MRI示:雙側基底節區、雙大腦腳斑片狀等T1短T2信號,液體衰減反轉恢復像(FLAIR)呈低信號;雙側丘腦、橋腦、雙額葉、左頂葉見斑片狀等T1長T2異常信號,FLAIR呈高信號。磁共振擴散加權成像(DWI)示:丘腦區病變呈高信號,雙側額葉見腦回樣高信號(右側較明顯),增強后雙側額葉輕度腦回樣強化,見圖2。
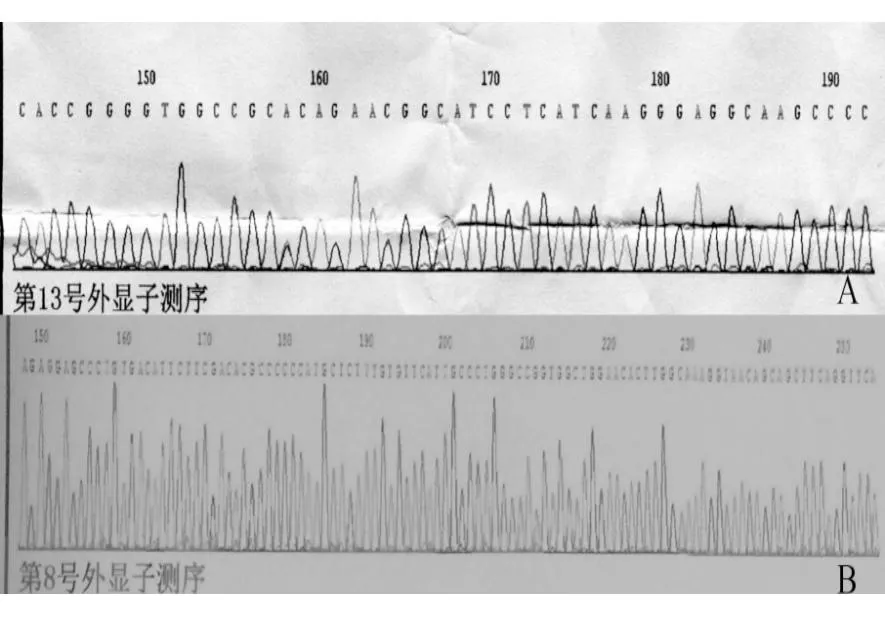
注:A:ATP7B基因第13號外顯子Ala1003Ala雜合同義突變; B:ATP7B基因第8號外顯子Arg778Leu錯義突變和Leu770Leu同義突變
圖1患者ATP7B基因檢測報告
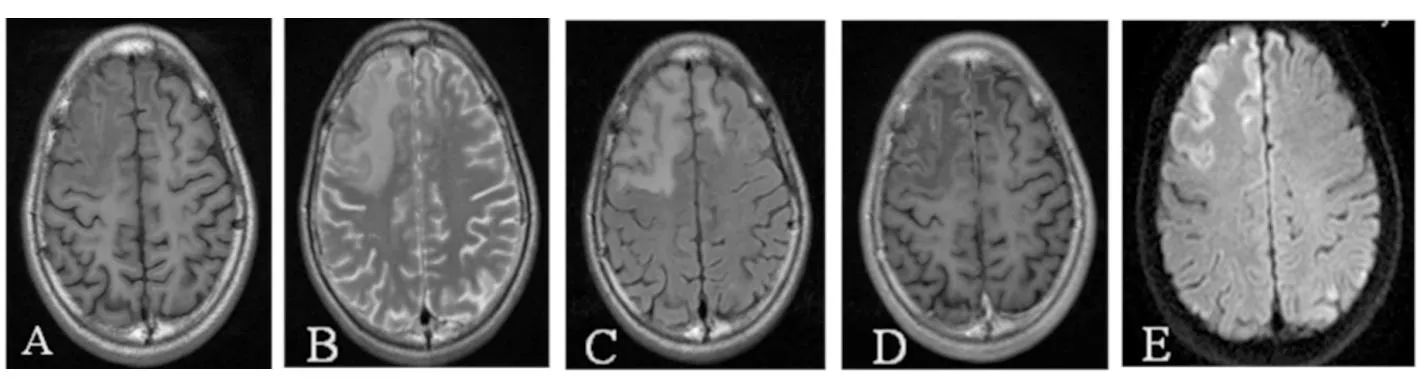
圖2 病例1頭顱MRI表現 雙側額葉等T1長T2異常信號影,Flair高信號,增強后見腦回強化,DWI見高信號
病例2,男性,47歲,以“動作緩慢、行走難以控制1年”于2013年8月30日入院治療。患者1年前出現步行時易跌倒、自覺步行時步速不自主加快,且無法控制易向前跌倒,當時未重視;自覺語速減慢,進食、進水偶有嗆咳;后逐漸出現手拿物品時手抖癥狀、無法手持物品,左手較右手嚴重。既往史及家族史無特殊。神經系統查體:神志清楚,時間、地點、人物定向可,言語清晰,語速慢;K-F環(+);四肢肌張力略高,肌力V級,雙側掌頦反射(+),左側Babinski征(+);行走緩慢,左上肢連帶差,行走時伴不自主顫動。生化檢查:血銅藍蛋白<0.1 g/L,24 h尿銅6.23 μmol。基因檢測:ATP7B基因第8號外顯子Arg778Leu錯義突變和Leu770Leu同義突變,見圖1B。腹部超聲提示肝硬化,肝區回聲增強增粗呈結節狀。頭顱MRI示:腦干片狀異常信號影,T1呈低信號,T2及FLAIR呈高信號,DWI呈等信號;雙側基底節區、側腦室旁及大腦皮層下多發斑片狀異常信號影,T1呈低信號,T2及FLAIR呈高信號,DWI增強后未見明顯異常強化灶。
2討論
本文2例WD患者的臨床特點為:(1)均以錐體外系癥狀起病,呈慢性進行性加重;(2)頭顱MRI表現非典型,表現為皮質下白質片狀或斑點低T1、高T2信號;(3)雙眼K-F征陽性;(4)生化檢查提示低銅藍蛋白血癥;(5)基因檢測提示ATP7B基因突變。本文2例患者頭顱MRI均表現為片狀皮質下損害,其中病例1患者主要累及額葉下大片白質。以皮質及皮質下損害的WD較為少見,這是一種特殊亞型[2],其具體病理特點暫未明確,可能與白質內銅沉積相關[3]。
一項納入100例WD患者的研究[4]發現,病變部位在殼核者占72%,尾狀核61%,丘腦58%,中腦49%,腦橋20%,大腦白質25%,皮質9%,延髓12%,小腦10%;且顱內MRI異常表現和患者臨床特點密切相關。本文復習了包括本研究在內的8例病例報告(共13例患者)的WD患者的臨床特點(表1),其中6例患者的頭顱MRI提示皮質片狀低T1、高T2信號,部分患者伴有異常信號強化和腦萎縮;其中1例患者頭顱CT見白質大片低密度灶,病理提示白質壞死。13例患者中,4例有基因診斷,均為ATP7B基因突變,但突變位點均不相同(表2)。ATP7B包含21個外顯子,不同人種有不同的突變熱點。研究[5]認為,不同位點突變和WD臨床癥狀相關。
典型WD神經精神系統表現主要包括動作遲緩、震顫、強直,精神癥狀、癡呆等,少有癲癇發作。13例患者中,表現為智力減退或癡呆6例,癲癇發作9例,表現為運動遲緩、活動障礙等11例。此類患者癲癇發作比例較高可能和病灶累及皮質及皮質下白質相關。1項包括490例WD患者的研究[6]有類似的結論,其中出現白質病灶的患者較無白質病灶的患者癲癇發生率更高。也有研究[3, 7]提示,患者出現癲癇發作后才發現顱內皮質及皮質下白質損害,但未明確兩者之間的因果關系。本研究中的2例患者未出現癲癇發作,需在今后隨訪中繼續觀察。
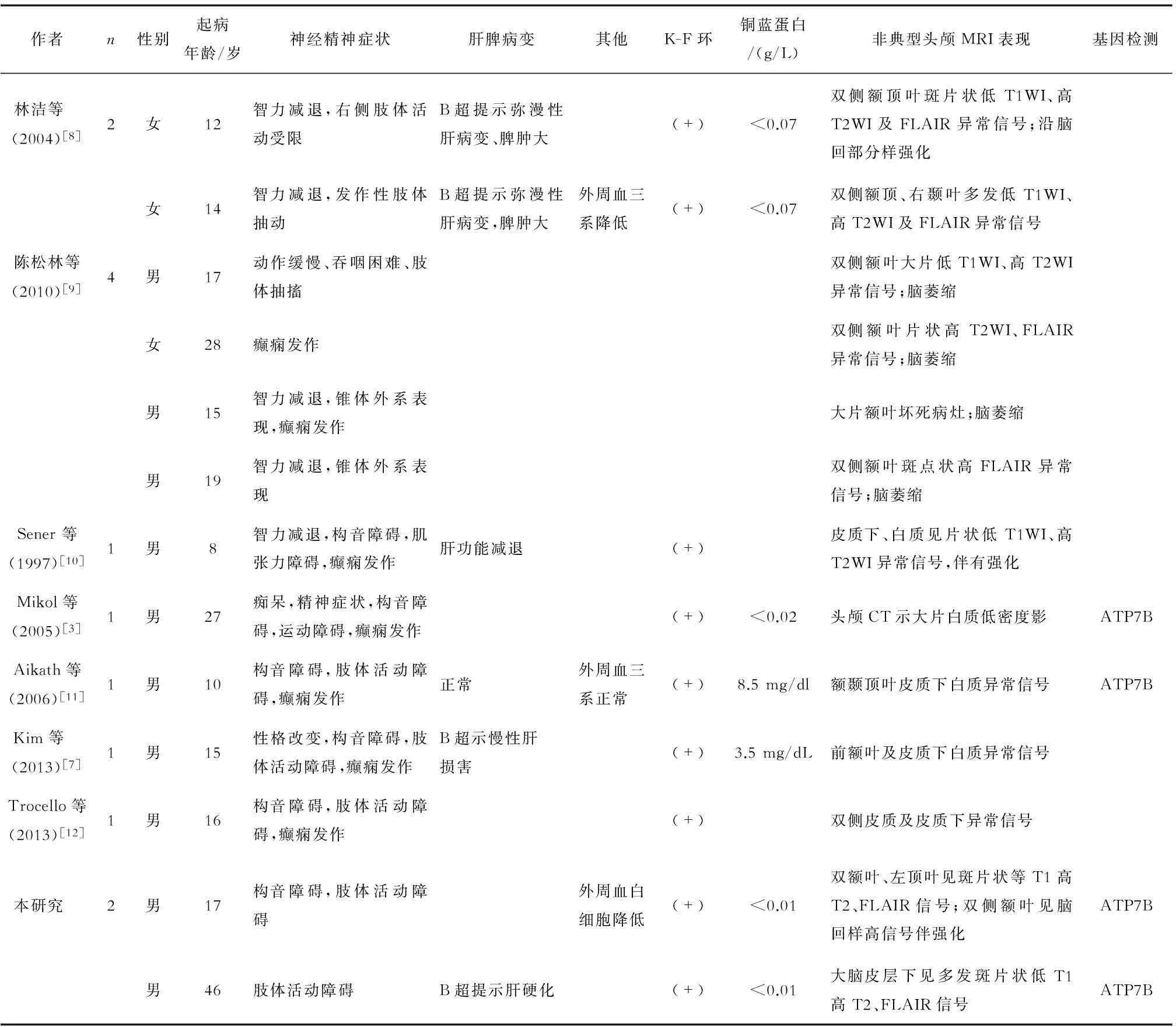
表1 非典型頭顱MRI表現的WD患者的文獻復習

表2 非典型頭顱MRI表現的WD患者的基因檢測
參考文獻
[1]Wilson S.Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver[J].Brain,1912,34:295-509.
[2]Finlayson MH,Superville B.Distribution of cerebral lesions in acquired hepatocerebral degeneration[J].Brain,1981,104(Pt 1):79-95.
[3]Mikol J, Vital C, Wassef M, et al.Extensive cortico-subcortical lesions in Wilson's disease: clinico-pathological study of two cases[J].Acta Neuropathol,2005,110(5):451-458.
[4]Sinha S, Taly AB, Ravishankar S, et al.Wilson's disease: cranial MRI observations and clinical correlation[J].Neuroradiology,2006,48(9):613-621.
[5]Li XH, Lu Y, Ling Y, et al.Clinical and molecular characterization of Wilson's disease in China: identification of 14 novel mutations[J].BMC Med Genet,2011,12:6.
[6]Prashanth LK, Sinha S, Taly AB, et al.Spectrum of epilepsy in Wilson's disease with electroencephalographic, MR imaging and pathological correlates[J]. J Neurol SciI,2010,291(1-2):44-51.
[7]Kim YE, Yun JY, Yang HJ, et al.Unusual epileptic deterioration and extensive white matter lesion during treatment in Wilson's disease[J].BMC Neurol,2013,13:127.
[8]林潔, 趙重波,呂傳真.肝豆狀核變性的磁共振成像特殊改變二例報告[J].中華神經科雜志,2005,38(3):186.
[9]陳松林, 梁穎茵, 周香雪,等.4例肝豆狀核變性頭顱核磁共振成像的非典型表現[J].世界臨床藥物,2010,31(5):291-294.
[10]Sener RN.MR imaging of Wilson's disease: contrast enhancement of the cerebral cortex, and corticomedullary junction[J].Comput Med Imaging Graph,1997,21(3):195-200.
[11]Aikath D, Gupta A, Chattopadhyay I, et al.Subcortical white matter abnormalities related to drug resistance in Wilson disease[J].Neurology,2006,67(5):878-880.
[12]Trocello JM, Woimant F, El Balkhi S, et al.Extensive striatal, cortical, and white matter brain MRI abnormalities in Wilson disease[J].Neurology,2013,81(17):1557.
Atypical Cranial MRI Manifestation of Wilson’s Disease: Two Case Reports and Review of Literatures
YUZhemingZHENGXiaominMAYuJINLirongDepartmentofNeurology,ZhongshanHospital,FudanUniversity,Shanghai200032,China
AbstractObjective:To explore the atypical cranial MRI manifestation of patients with Wilson’s disease (WD) . Methods: The clinical features and cranial MRI manifestation of two patients with WD, who were confirmed with clinical biochemistry and gene diagnosis, were analyzed. Retrospectively analysis of reported cases was conducted based on review of literatures. Results: The typical WD MRI manifestations were lesions at bilateral lenticular nucleus, thalamus and midbrain. However, these two cases showed subcortical hypointense on T1WI, hyperintense on T2WI, and Flair hyperintense, which were rare. According to the literature, 13 WD patients’ cranial MRI showed abnormal signal of subcortical white, and lesions involved cerebral cortex in 9 of them(9 cases of frontal lobe, 4 cases of parietal lobe, 2 cases of temporal lobe ). Among these 13 patients, 9 contained extrapyramidal manifestation as bradykinesia, movement disorder and etc, 6 had intellectual impairment or dementia, and 9 had epileptic seizure. The high rate of seizure might be related to cortical or subcortical lesions, which had no special relationship with particular mutation in ATP7B gene. Conclusions: The cranial MRI manifestations of WD patients are variable, and closely related to clinical symptoms.
Key WordsHepatolenticular degeneration;MRI;Gene
中圖分類號R742.4
文獻標識碼A
猜你喜歡
中國當代醫藥(2016年30期)2017-01-07 13:03:05
中國實用醫藥(2016年30期)2016-12-28 14:58:58
中國實用醫藥(2016年29期)2016-12-26 10:14:12
心腦血管病防治(2016年5期)2016-12-19 07:30:05
華夏醫學(2016年4期)2016-12-12 00:49:41
中國實用醫藥(2016年28期)2016-12-07 22:14:45
中國現代醫生(2016年23期)2016-11-15 03:35:37
科技視界(2016年18期)2016-11-03 20:32:54
中國實用醫藥(2016年21期)2016-08-19 12:42:42
中國實用醫藥(2016年22期)2016-08-19 12:11:18
