功能化離子液體雜多酸鹽催化丁二酸銨水溶液直接酯化
2015-06-19 07:52:28張鋒梁金花胡曦王俊格陳劉龍任曉乾姜岷
化工學報 2015年7期
關鍵詞:催化劑
張鋒,梁金花,胡曦,王俊格,陳劉龍,任曉乾,姜岷
(1南京工業大學化學化工學院,江蘇 南京210009;2南京工業大學生物與制藥工程學院,江蘇 南京 211816)
引 言
丁二酸是生物煉制產品中重要的 C4平臺化合物。微生物發酵法利用葡萄糖、秸稈等再生生物質以及CO2為原料生產丁二酸,其污染少、成本低、生產環境良好,近年來成為國內外研究的熱點[1-3]。但發酵液中丁二酸的濃度低,且存在菌體、色素和無機鹽等雜質,因此提取純化丁二酸成為發酵法生產丁二酸的關鍵問題。文獻報道有關發酵液中提取丁二酸的方法有鈣鹽與銨鹽法[4-5]、膜分離法[6-7]、溶劑萃取法[8-9]、結晶法[10]、酯化水解法[11]。與能耗高、工藝復雜、酸堿用量大的傳統工藝相比,直接酯化水解法是一種更加高效的生物基羧酸提取方法[12-14]。直接酯化水解法是將發酵液中的羧酸或羧酸鹽(如羧酸銨)與醇直接進行酯化,分離出酯,然后水解羧酸酯得到高純度羧酸,同時酯化劑醇和由銨鹽產生的氨氣也可循環使用(圖 1),具有能耗低、工藝簡單的優點。
直接酯化水解法提取生物基丁二酸的關鍵是丁二酸(或丁二酸鹽)水溶液的直接酯化過程以及高效易回收催化劑的應用。目前,國內外有關水溶液體系中丁二酸(或丁二酸鹽)直接酯化的文獻報道較少并且也存在著一定的問題。Budarin等[15]制備了磺化型活性炭(Starbon?)催化劑,成功應用于 6%(質量分數)丁二酸水溶液與乙醇的直接催化酯化,酯化率可達90%,但催化劑制備條件要求較高、時間長,過程較復雜;Delhomme 等[16]分別將十二烷基苯磺酸、樹脂催化劑(Nafion?NR-50)和脂肪酶催化劑(Novozym 435)應用于催化丁二酸水溶液與正辛醇的酯化反應,3種催化劑均可取得較好的酯收率,但同時也指出十二烷基苯磺酸易乳化,難于從反應體系中分離,Nafion?NR-50酯收率達90%所用時間較長(72h),Novozym 435價格較高;張英偉等[17]利用高濃度的丁二酸銨溶液(70%~90%,質量分數)在加壓條件下與甲醇或乙醇進行反應,酯化收率可達98%,但其工藝路線較長,反應條件要求較高。因此,選擇高效合理的催化劑以及優化工藝條件在酯化水解法提取生物基丁二酸過程中顯得至關重要。
離子液體應用于催化酯化反應,與傳統酯化催化劑濃硫酸、固體超強酸、強酸性陽離子交換樹脂、雜多酸等相比,在催化性能與環境保護方面有更多的優勢[18-19],其應用也越來越廣泛。趙地順等[20-21]將酸性室溫離子液體應用于催化固體丁二酸的酯化,得到了較好的酯收率,離子液體表現出良好的催化活性。基于離子液體的液-固非均相催化體系研究,利用離子液體分子結構和酸性具有可調控的特性,Leng等[22-23]將功能化的離子液體有機陽離子與具有Keggin結構的雜多酸陰離子結合,制備出一種新型的固體有機-無機雜化材料,其熔點高于100℃,在很多酯化反應中表現出“高溫溶解均相催化,低溫自相分離析出”的特性,而且催化劑用量很少,兼備均相催化劑與非均相催化劑的優點。這種特性既可以在反應中獲得高效的催化性能又可簡化催化劑的分離回收過程。而將此類催化劑應用于催化水溶液體系中丁二酸(或丁二酸鹽)的直接酯化,尚未見相關報道。
發酵法制備丁二酸的過程中,為了消除生成的丁二酸對大腸桿菌活性的反饋抑制,往往加入氨水來調節pH,得到丁二酸銨水溶液。直接酯化法中將其濃縮到一定濃度,再用醇進行酯化。本文重點探討丁二酸銨水溶液的直接酯化,合成一系列磺酸功能化離子液體雜多酸鹽作為反應催化劑,以正丁醇為反應酯化劑。篩選出催化性能最好的雜多酸鹽,進一步探討并確定丁二酸銨水溶液直接酯化反應的最佳實驗工藝條件。
1 實驗部分
1.1 主要試劑
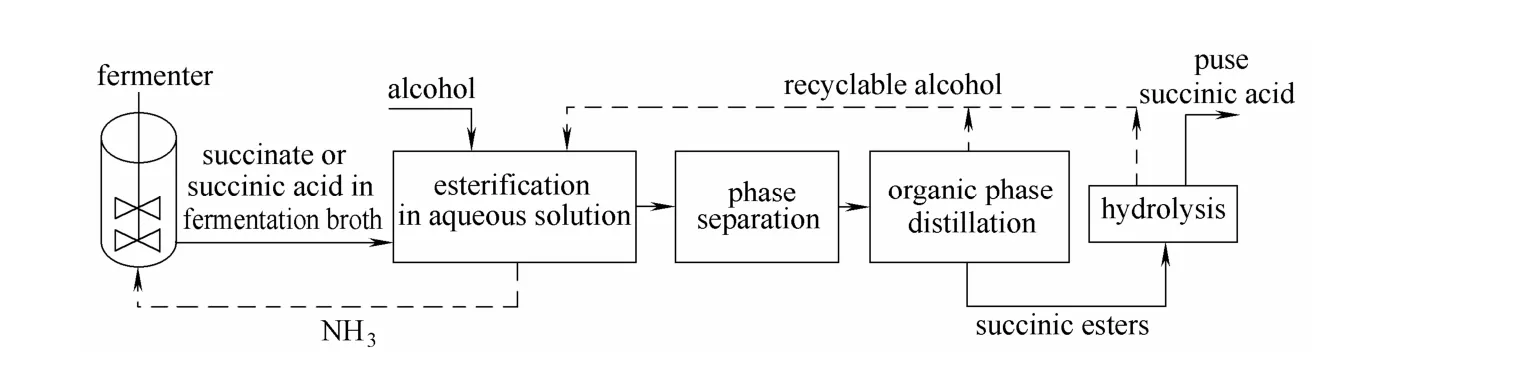
圖1 酯化水解法的工藝過程Fig.1 Process of esterification and hydrolysis method
試劑:丁二酸、N-甲基咪唑、氯仿、乙醚、吡啶、三乙胺、正丁醇、對硝基苯胺、氫氧化鈉、丁二酸二丁酯、丁二酸單丁酯、異辛醇、硅鎢酸、磷鎢酸、磷鉬酸、濃硫酸均為分析純,國藥集團化學試劑有限公司;1,3-丙磺酸內酯,分析純,江蘇夢得電鍍化學品有限公司;Nafion?NR-50,Aldrich化學試劑公司;[MIMPS]HSO4和30%~70%(質量分數)丁二酸銨水溶液,實驗室自制。
1.2 催化劑的合成
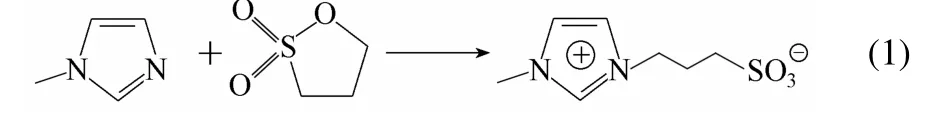
催化劑合成主要分兩步進行(以[MIMPS]3PMo12O40的合成為例):將1,3-丙烷磺酸內酯(0.10 mol)、50 ml氯仿加入三口燒瓶中,冰水浴,攪拌,向燒瓶中緩慢滴加N-甲基咪唑(0.10 mol),完畢,室溫下反應6 h,過濾。所得白色固體用乙醚洗滌3次,得1-丙基磺酸基-3-甲基咪唑鹽(MIMPS),其合成過程見式(1)。同樣方法制得磺酸基吡啶鹽(PYPS)、磺酸基三乙胺鹽(TEAPS)。
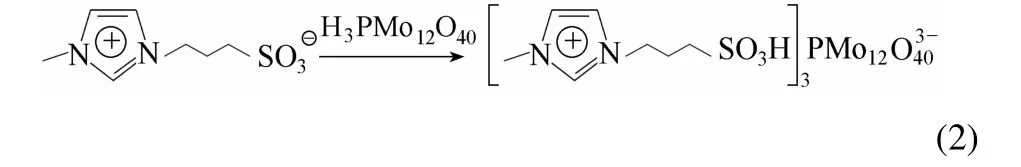
將0.01 mol的磷鉬酸加入100 ml去離子水中,得磷鉬酸水溶液,再加入0.03 mol MIMPS,攪拌,室溫下反應24 h。反應結束,混合物經旋蒸除去水分,所得固體用乙醚洗3次,80℃真空干燥12 h,即制得磺酸功能化離子液體磷鉬酸鹽催化劑([MIMPS]3PMo12O40),合成過程如下
同理,[MIMPS]4SiW12O40、[MIMPS]3PW12O40、[PYPS]3PMo12O40、[PYPS]3PW12O40、[PYPS]4SiW12O40、[TEAPS]3PW12O40、[TEAPS]4SiW12O4、[TEAPS]3PMo12O40、[MIM]3PMo12O40、[TEA]3PMo12O40和[PY]3PMo12O40的合成方法如上。
1.3 催化劑的表征
采用Thermo Nicolet公司的NEXUS系列智能型傅里葉紅外光譜儀對合成的催化劑進行FT-IR表征,測試條件為:KBr壓片,波數范圍為4000~400 cm?1,分辨率 4 cm?1,掃描次數 32次;采用美國Perkin Elmer公司的PerkinElmer 2400型元素分析儀對催化劑進行CHN元素分析;采用Hammett指示劑對硝基苯胺(pKa=0.99)與紫外-可見光譜聯用法測定催化劑的酸強度,在美國 PerkinElmer公司生產的 LAMBDA950型紫外-可見光譜儀上進行,掃描范圍200~800 nm;采用上海儀電科學儀器股份有限公司生產的ZJD-4A型自動電位滴定儀對離子液體的酸量進行分析,用0.01 mol·L?1的NaOH水溶液滴定。
1.4 酯化反應
反應在帶有溫度計、油水分離器、回流裝置與真空泵的三口燒瓶中進行。向燒瓶中加入 15.21 g(0.1 mol)丁二酸銨,再加15.21 g去離子水,使其溶解;再依次加入37.06 g正丁醇和0.15 g 催化劑;確保整個反應裝置體系氣密性良好,磁力攪拌,加熱溫度為130℃,打開真空泵(真空壓力為0.02 MPa)。一定時間后停止反應,靜置冷卻至室溫,分析產物。篩選出最佳催化劑,分別考察催化劑用量、醇銨摩爾比、丁二酸銨溶液濃度、反應溫度、反應時間和真空度對酯化反應的影響。
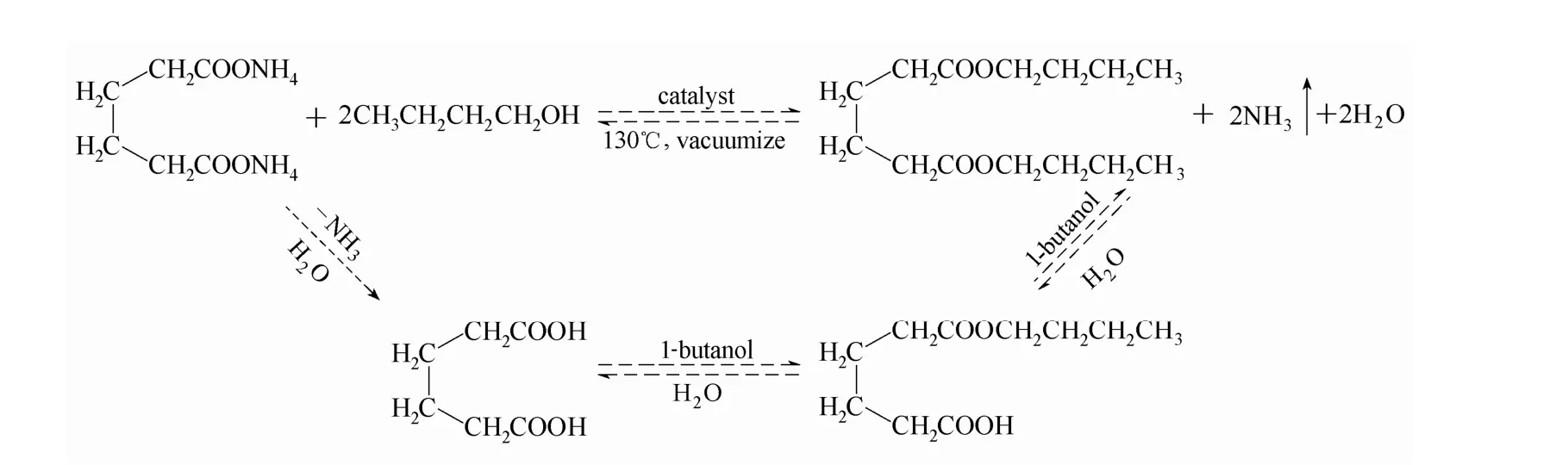
圖2 丁二酸銨直接酯化反應過程Fig.2 Esterification reaction process of ammonium succinate
丁二酸銨直接酯化反應過程如圖2所示。加熱條件下,水溶液中丁二酸銨受熱分解生成氨氣和丁二酸;氨氣被真空泵及時抽出,促使反應向右進行,不斷生成丁二酸;催化劑催化丁二酸與正丁醇酯化生成丁二酸單丁酯,單丁酯進一步與正丁醇反應生成雙酯。反應過程中正丁醇既作酯化劑又作帶水劑,正丁醇與水形成共沸物,經油水分離器不斷將水帶出反應體系,從而相對增加了反應物濃度,又由酯化反應可逆的特性,水的移出促使反應不斷向右移動,從而加快反應進程。
1.5 產物分析
反應產物由 HP-4890D型氣相色譜儀進行分析,采用SE-54型毛細管柱(柱長30 m,內徑0.32 mm,膜厚1.5 μm),FID檢測器。分析條件:柱溫初始為100℃,以15℃·min?1程序升溫至220℃,檢測時間為15 min;檢測器溫度280℃;汽化室溫度280℃;高純氮氣作載氣;進樣量為0.2 μl。以異辛醇為內標物,采用內標法分析產物樣品中單、雙酯的含量,進一步計算得到單、雙酯的量。酯收率為丁二酸單丁酯收率與丁二酸二丁酯收率之和,按式(3)計算。
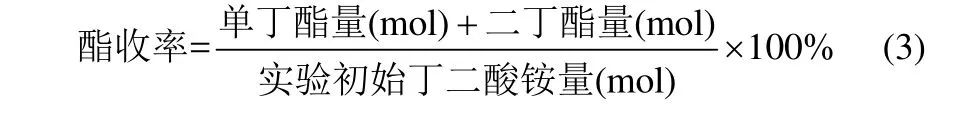
2 結果與討論
2.1 催化劑的表征
2.1.1 CHN元素分析 由表1元素分析結果可以看出,所合成催化劑的 C、H、N元素含量測量值與理論值基本一致。

表1 C、H、N元素分析結果Table 1 Results of C、H、N elemental analysis /%(mass)
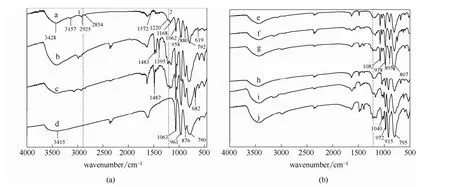
圖3 催化劑的紅外圖譜Fig.3 FT-IR spectra of catalysts
2.1.2 催化劑的 FT-IR 與相關文獻中紅外圖譜進行分析對比,相同基團所對應的特征峰位置基本一致。圖中譜線a中3157、619、1572 cm?1處的峰分別對應咪唑環中 N—H、C—H、鍵的伸縮振動;譜線b中1483、1395 cm?1處的峰分別對應三乙胺中C—C、C—H鍵的伸縮振動;譜線c中1487、682 cm?1處的峰分別為吡啶環中、C—H鍵的伸縮振動[24-25];譜線a中1168、1220 cm?1處的峰對應磺酸基中鍵的伸縮振動(圖中虛線2處),2925、2854 cm?1處的峰對應丙基中飽和C—H鍵的伸縮振動[23,26](圖中虛線1處),由此說明催化劑中存在[MIMPS]+、[PyPS]+、[TEAPS]+。另外,催化劑仍然保留了雜多酸的Keggin結構:如譜線 a中1062、958、880和792 cm?1分別對應磷鉬酸根中P—Oa、、Mo—Ob—Mo、Mo—Oc—Mo 鍵的伸縮振動(其中880 cm?1對應共角八面體之間的橋鍵,792 cm?1對應共邊八面體之間的橋鍵);如譜線g中1082、978、895、807 cm?1分別對應磷鎢酸根中P—Oa、、W—Ob—W、W—Oc—W鍵的伸縮振動;如譜線 j中 1040、972、915、795 cm?1分別對應磷鎢酸根中Si—Oa、、W—Ob—W、W—Oc—W鍵的伸縮振動[25-26]。由圖3(a)還可以看出,與譜線 d純磷鉬酸相比,[MIMPS]3PMo12O40中Keggin結構的特征峰值都發生了一定的偏移,而且雜多酸表面的O—H振動峰值由3415 cm?1偏移至3428 cm?1,這均是功能化陽離子與磷鉬酸根結合相互作用的結果[23]。由上述催化劑的 IR表征以及結合表1的C、H、N元素分析結果可知,所合成的離子液體雜多酸鹽與設計結構基本一致。
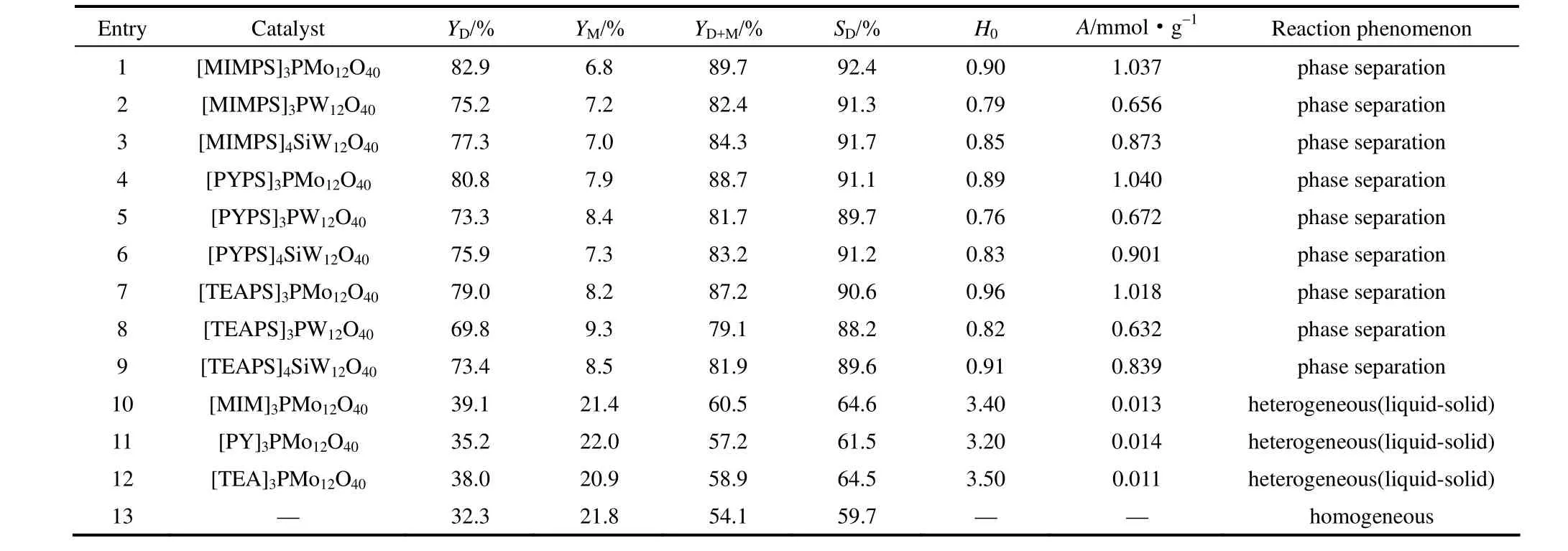
表2 不同離子液體雜多酸鹽(等質量)催化性能的比較Table 2 Catalytic performance of different functionalized heteropolyacid salts (same mass)
2.2 催化劑的性能比較
2.2.1 合成催化劑性能比較 考察所合成的一系列離子液體雜多酸鹽的催化性能,實驗結果見表 2、表3。
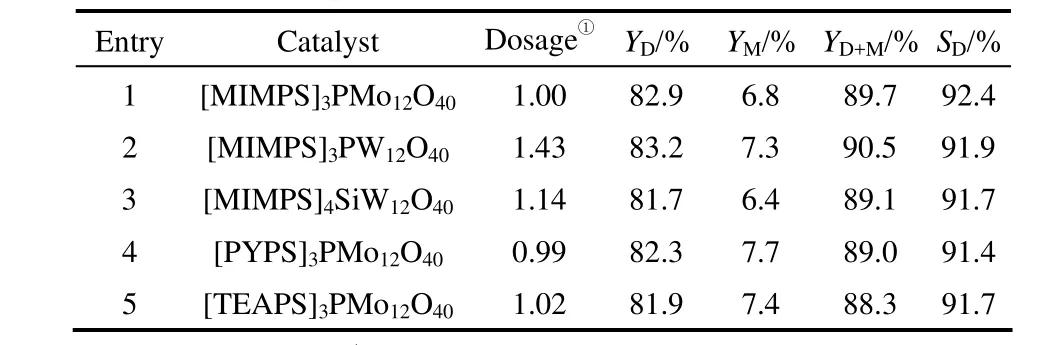
表3 不同離子液體雜多酸鹽(含等物質的量的PS)催化性能的比較Table 3 Catalytic performance of different functionalized heteropolyacid salts (same molar of PS)
由表2中實驗現象可以看出,無磺酸基團(PS)的[MIM]3PMo12O40、[PY]3PMo12O40、[PY]3PMo12O40表現為液-固非均相催化,而磺酸功能化離子液體雜多酸鹽在本反應中表現出“高溫溶解均相催化,低溫自相分離析出”的自分離特性。這是由于陰陽離子間的氫鍵作用以及結構中具有較大體積的高價雜多陰離子使得這類離子化合物具有較高的熔點(>100℃),常溫下為固體[22];又由于磺酸基團(PS)的引入使其具有受熱易溶解的性質,在本反應過程中與有機體系成均相,從而進行均相催化;反應結束后,溫度下降催化劑從反應體系中沉淀析出。等質量催化劑條件下,表2中entries 1~9催化劑的酯收率均在80%以上,丁二酸二丁酯的選擇性在90%以上,而entries 10~12催化劑的酯收率及丁二酸二丁酯選擇性僅在60%左右,與未加催化劑時相差較小,這說明磺酸基團的引入提高了催化劑的活性。另外,對比entries 1~9催化劑,當功能化有機陽離子(XPS)相同時,催化劑催化活性表現為[XPS]3PMo12O40> [XPS]4SiW12O40> [XPS]3PW12O40,這與表中催化劑的酸強度順序不一致,而與其酸量大小順序一致,這可能是由于相同質量條件下,酸量對反應的影響較大,磺酸根量與催化劑酸量呈正比關系,的分子量遠小于 PW12和SiW12,所含磺酸根量相對較多,因此催化劑具有更多的酸位中心,進而提高了其催化活性;而雜多酸陰離子(Y)相同時,催化活性[MIMPS]3Y >[PYPS]3Y > [TEAPS]3Y,這可能是由于陰離子與不同的陽離子形成的特定的網絡結構所決定的,[MIMPS]3Y在溶劑中更能形成氫鍵,與反應物相溶性更好[23],但三者催化活性相差很小,這可能是由于三者的酸強度及酸量相差較小。
當催化劑含等物質的量的磺酸基團時,實驗結果見表3。entries 1~5催化劑的酯收率及選擇性基本相同,這可能是由于此時幾種催化劑具有相同的酸量,又由表2中可以看出其酸強度相差不大。又對比表3中含不同雜多酸陰離子的entries 1~3催化劑:催化劑用量順序為[MIMPS]3PW12O40>[MIMPS]4SiW12O40> [MIMPS]3PMo12O40;對比含不同有機陽離子的entries 1、4、5,3種催化劑用量相差很小,這均與表2的分析結果相對應,這也進一步說明了本反應中磺酸基團是影響催化劑活性的主要因素,陰陽離子本身對催化劑活性的影響較小。綜上,催化劑中雜多陰離子使其具有較高的熔點,磺酸功能化基團(PS)使其具有很高的催化活性和較好的溶解性;催化劑相同用量下,[MIMPS]3PMo12O40、[PYPS]3PMo12O40和[TEAPS]3PMo12O40用于丁二酸銨水溶液直接酯化所得酯收率及選擇性較高,為本反應理想催化劑。
2.2.2 與傳統催化劑的性能比較 將上述具有最好催化活性的[MIMPS]3PMo12O40與幾種傳統酯化催化劑在相同的實驗條件下進行比較。分別考察各催化劑對丁二酸銨水溶液直接酯化的催化性能,結果見表4。
由表4可知,[MIMPS]3PMo12O40酸強度與其他幾種傳統催化劑(entries 2~5)相差不大,但應用于本反應中其催化性能存在較大差別。對比[MIMPS]3PMo12O40,純雜多酸催化劑(entries 2~4)也可取得較好的酯收率和選擇性,但由于具有良好的溶解性使其難以從反應體系中分離;傳統室溫離子液體[MIMPS]HSO4及 H2SO4的催化活性均比[MIMPS]3PMo12O40低,這可能是由于其過多的 H+易與丁二酸銨熱解產生的NH3結合再次形成NH+4,從而影響NH3的及時排出,不利于反應向正方向進行;Nafion?NR-50是一種固體超強酸,但在反應中催化活性較差,這可能是由于作為非均相催化劑,反應中存在較大的傳質阻力。綜上,磺酸功能化離子液體雜多酸鹽兼備均相催化劑與多相催化劑的優點,與幾種傳統酯化催化劑相比,在反應中表現出更好的催化性能,因此選 [MIMPS]3PMo12O40作為本反應最理想的催化劑,進一步探討丁二酸銨水溶液直接酯化的最佳反應工藝條件。
2.3 反應條件的確定
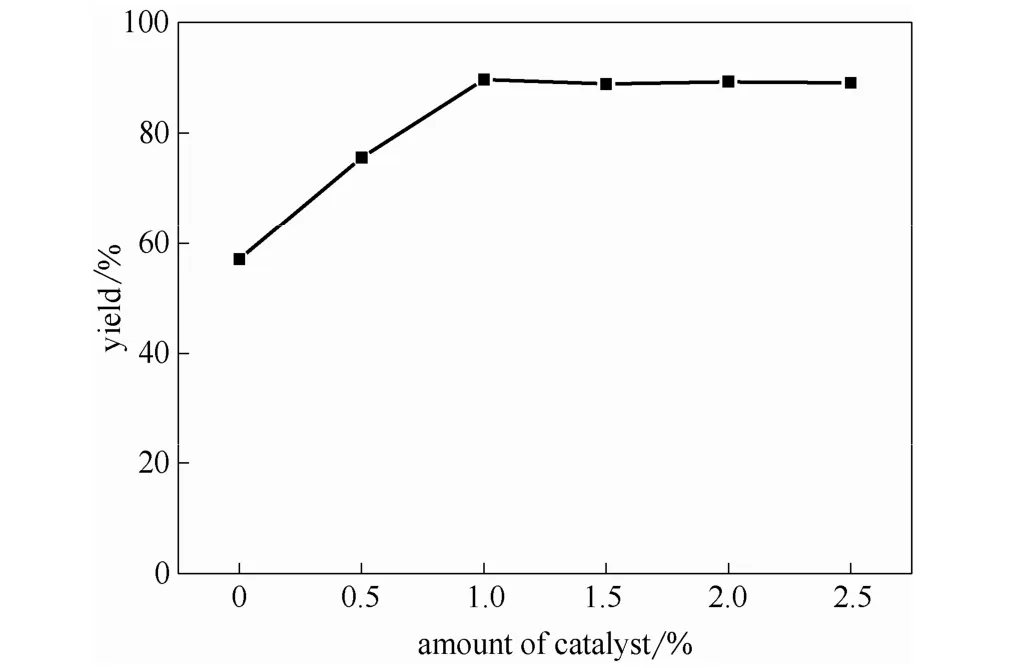
圖4 催化劑用量對酯收率的影響Fig.4 Effects of catalyst dosage on esterification yield
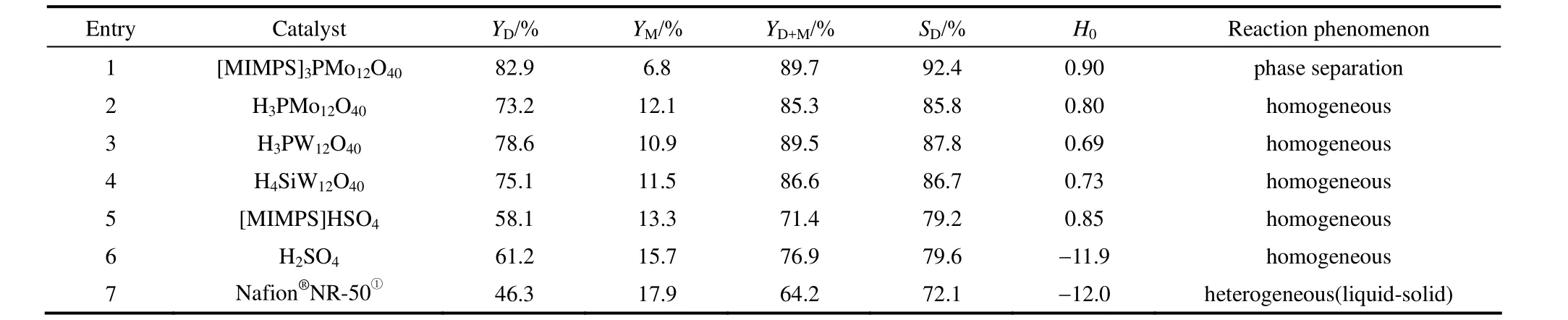
表4 不同催化劑催化性能以及酸性比較Table 4 Comparison of catalytic performance and acidity of different catalysts
2.3.1 催化劑用量的影響 催化劑用量是影響酯收率的一個重要因素,其用量大小直接影響催化反應速率的快慢。一定范圍內增加催化劑用量,催化活性中心數目增加,有利于酯化反應的進行。在醇銨摩爾比為 5:1,丁二酸銨質量分數為 50%,反應溫度為130℃,反應時間為12 h,真空度為0.02 MPa的條件下考察催化劑用量(0~2.5%,質量分數)對酯收率的影響,實驗結果如圖4所示。由圖可以看出,在催化劑用量為 0~1.0%時,酯收率隨催化劑用量的增加而增加,在1.0%時達最大值89.7%,而繼續增加催化劑用量(>1.0%)時,丁二酸酯收率基本趨于穩定。因此,催化劑的最佳用量為 1.0%。[MIMPS]3PMo12O40用量很少,同時說明了其應用于本反應具有很好的催化活性。
2.3.2 醇銨摩爾比的影響 在催化劑用量為1.0%,丁二酸銨質量分數為50%,反應溫度為130℃,反應時間為12 h,真空度為0.02 MPa的條件下考察醇銨比(2:1~7:1)對酯收率的影響,實驗結果如圖5所示。由圖可以看出:醇銨摩爾比在2:1~5:1范圍內,丁二酸酯收率隨丁醇量的增加而增加,這是由于反應物正丁醇量的增加,使反應過程中反應物分子間的碰撞概率增大,使得酯化速率加快,同時正丁醇與水共沸,正丁醇又作為帶水劑不斷將水帶出反應體系,促進反應向正方向移動,從而提高酯收率。在醇銨比為5:1時,酯收率達最大值,當醇銨比超過 5:1,酯收率有下降趨勢。這可能是一方面由于過量的正丁醇相對降低了反應中丁二酸以及催化劑的濃度,另一方面過量正丁醇降低了反應體系溫度。因此,最佳醇銨摩爾比為5:1。
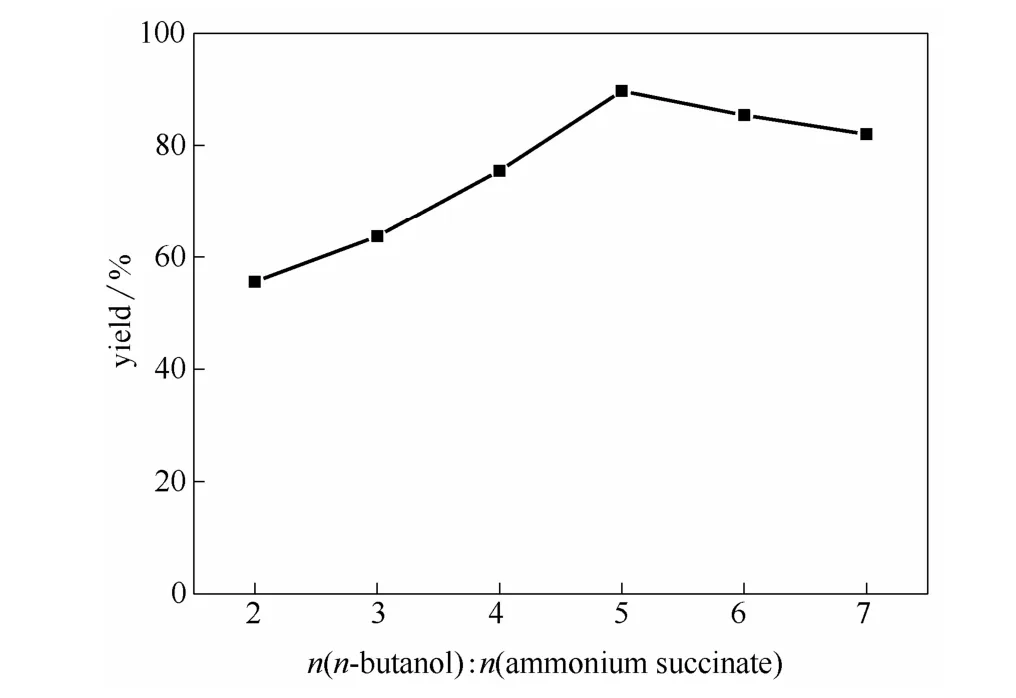
圖5 正丁醇與丁二酸銨摩爾比對酯收率的影響Fig.5 Effects of molar ratio of n-butanol to ammonium succinate on esterification yield
2.3.3 丁二酸銨水溶液濃度的影響 以[MIMPS]3PMo12O40為催化劑,在催化劑用量為1.0%,醇銨摩爾比為5:1,反應溫度為130℃,反應時間為12 h,真空度為0.02 MPa的條件下考察不同丁二酸銨濃度(30%~70%,質量分數)對酯收率的影響,結果如圖6所示。可以看出,在一定濃度范圍內,當丁二酸銨濃度增加時酯收率有所增加,但增加幅度不大。這是由于反應過程中,帶水劑正丁醇將丁二酸銨水溶液中的水帶出體系所用時間(10~30 min)相對總反應時間(12 h)較短,水被及時帶出,從而對實驗結果影響相對較小,但除水耗能較大。因此,從耗能和酯收率角度考慮,丁二酸銨最佳反應濃度為50%。
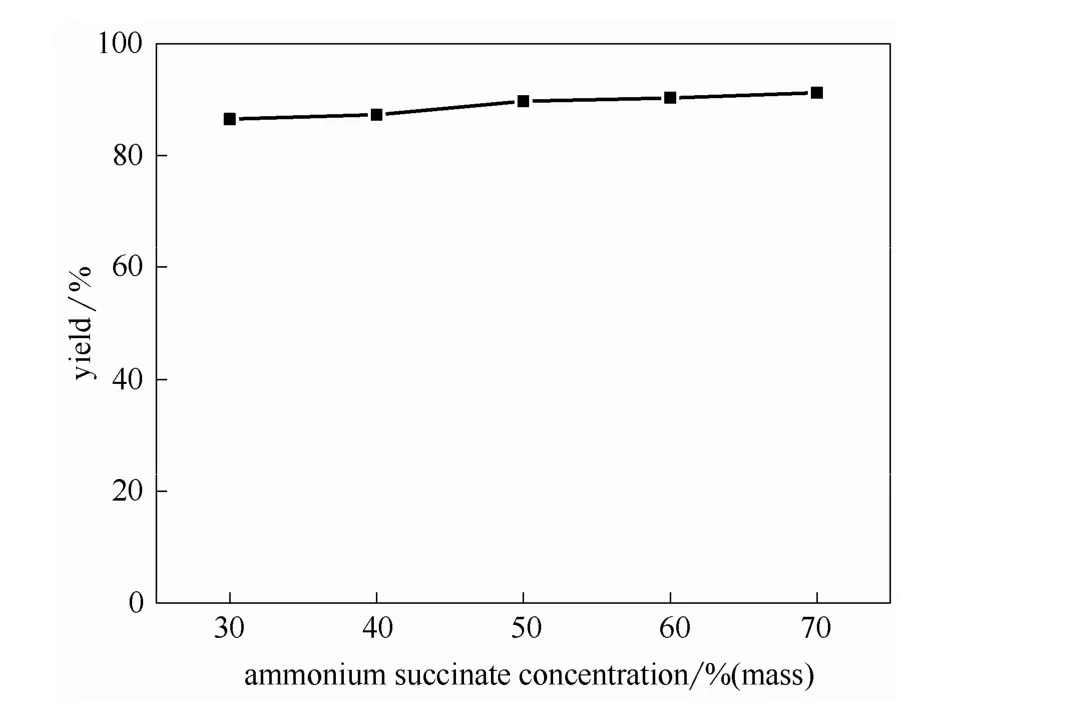
圖6 丁二酸銨濃度對酯收率的影響Fig.6 Effects of ammonium succinate concentration on esterification yield

圖7 反應溫度對酯收率的影響Fig.7 Effects of reaction temperature on esterification yield
2.3.4 反應溫度的影響 溫度是影響酯化反應的一個重要因素,適當升高溫度可加快反應速率,但過高的溫度也會引起一些副反應的發生。在催化劑用量為 1.0%,醇銨摩爾比為 5:1,丁二酸銨濃度為50%,反應時間為12 h,真空度為0.02 MPa的條件下,考察了溫度(110~150℃)對酯收率的影響,實驗結果如圖7所示。溫度較低時,丁二酸銨分解緩慢,影響酯化反應的進行,110℃時酯收率僅為40.7%。溫度在 110~130℃階段,隨溫度升高丁二酸銨分解加快,酯化速率加快,酯收率增加幅度加大,130℃時酯收率達89.7%。但溫度超過130℃后,收率持續下降,這主要是由于高溫下丁二酸銨自身的熱分解加劇,過多氨氣不能及時排除,高溫下易產生酰胺等副產物,從而導致酯收率降低。因此,最佳反應溫度為130℃。
2.3.5 反應時間的影響 在催化劑用量為1.0%,醇銨摩爾比為 5:1,丁二酸銨濃度為 50%,反應溫度為130℃,真空度為0.02 MPa的條件下考察了反應時間對酯收率的影響。實驗結果與圖2反應式相一致,如圖8所示,酯收率在3 h后有較大幅度的增加;反應時間3~8 h階段丁二酸單丁酯和丁二酸二丁酯收率都在增加,但單酯化率大于雙酯化率;在8~12 h階段,丁二酸單丁酯收率逐漸降低,而丁二酸二丁酯收率快速上升,這是由于該階段反應物丁二酸銨量的減少使得單酯化反應接近平衡,但丁二酸單丁酯積累量不斷增加,促使單丁酯進一步酯化成雙酯的速率加快;12 h后,單酯化反應基本平衡,單酯不斷酯化轉化為雙酯,但雙酯量增加緩慢,而總酯收率基本趨于穩定。因此,最佳反應時間為12 h。
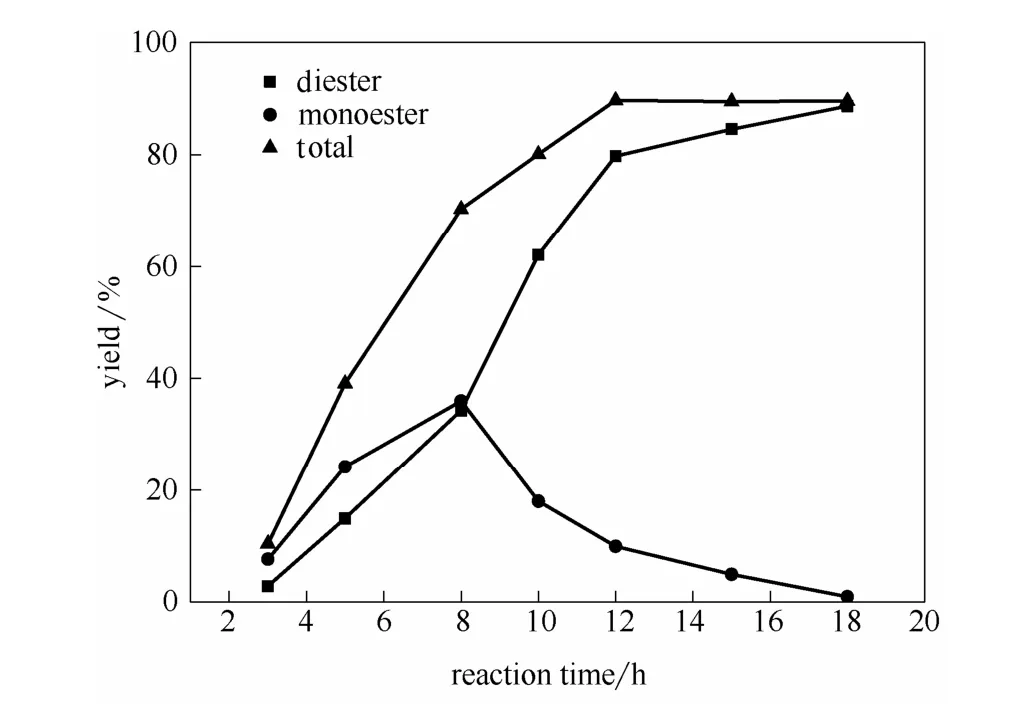
圖8 反應時間對酯收率的影響Fig. 8 Effects of reaction time on esterification yield
2.3.6 真空度的影響 丁二酸銨水溶液直接酯化合成的丁二酸酯,丁二酸銨首先熱解形成氨氣和丁二酸,將反應生成的氨氣及時用真空泵抽出,可促進反應向右進行,從而提高丁二酸酯的收率。在催化劑用量為1.0%,醇銨摩爾比為5:1,丁二酸銨質量分數為50%,反應溫度為130℃,反應時間為12 h的條件下考察真空度(0~0.4 MPa)對酯化反應的影響,結果如圖9所示。可以看出,開始一段范圍內隨著真空度的增加酯收率逐漸增加,在真空度為0.02 MPa時達最大值。當真空度大于0.02 MPa時,酯收率出現下降趨勢,這是由于真空度過大,部分正丁醇蒸氣不能及時冷卻回流而被抽出反應體系,使得酯化劑量不足。因此,最佳真空度為0.02 MPa。
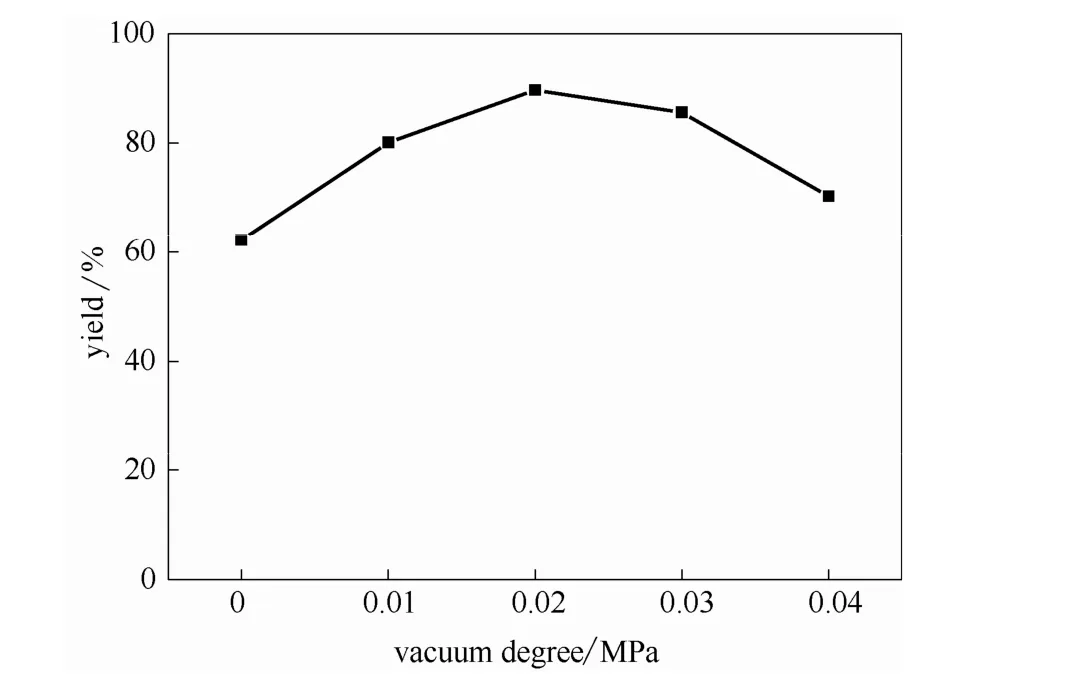
圖9 真空度對酯收率的影響Fig.9 Effects of vacuum degree on esterification yield
2.4 催化劑的重復使用性能
反應結束后,將靜置冷卻至室溫的液固混合物倒入離心管中離心;離心后將上層透明溶液倒出,80℃下減壓旋蒸,蒸出正丁醇,即得丁二酸酯;用丁醇將離心管底部的固體催化劑洗滌3次,真空干燥,準確稱量,計算催化劑的回收率(與初次加入的催化劑量比)。以[MIMPS]3PMo12O40作催化劑,其用量為1.0%,醇銨摩爾比為5:1,丁二酸銨濃度為50%,反應溫度為130℃,反應時間為12 h,真空度為 0.02 MPa的最優條件下考察催化劑的重復使用性能。重復性實驗結果如圖10所示。
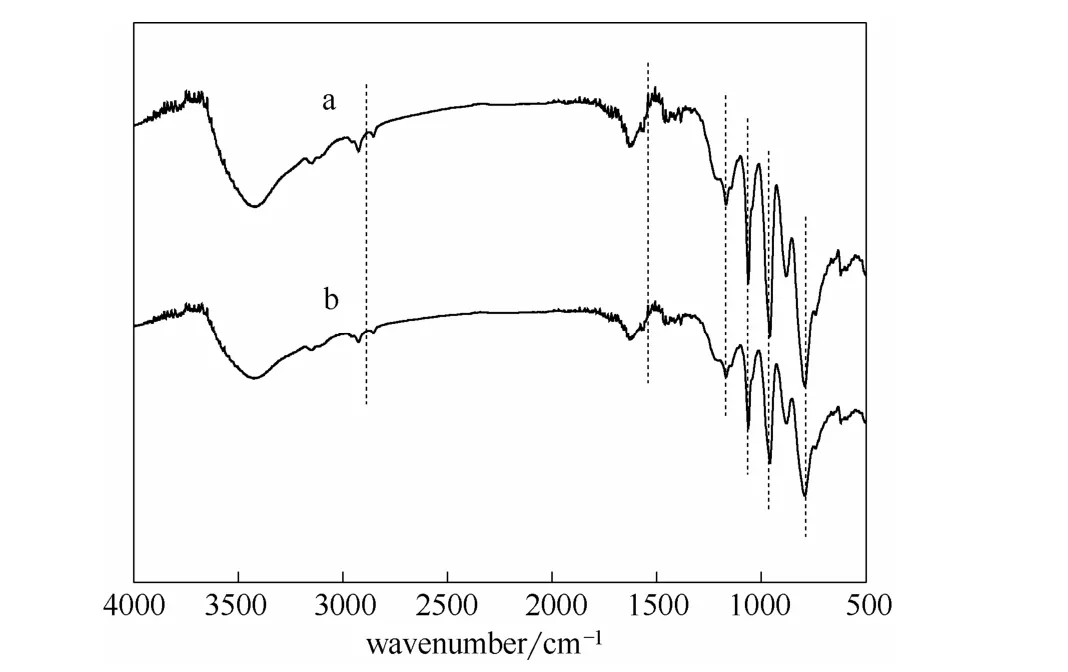
圖10 新鮮及回收催化劑的紅外圖譜Fig.10 FT-IR spectra of fresh catalyst(a)and recovered catalyst after 6 times reuse(b)
由表5可以看出,隨著催化劑使用次數增多,酯收率及丁二酸二酯選擇性均出現略微的下降,這可能是由于催化劑回收時有少部分質量損失,但使用多次后酯收率仍能保持 85%以上。圖 10中[MIMPS]3PMo12O40使用前后的兩個紅外譜圖基本一致(如圖中虛線處對應的特征峰位置),說明[MIMPS]3PMo12O40經多次使用后仍保留了原有結構。綜上說明該催化劑不僅具有很好的催化活性,而且具有較高的重復使用性能。
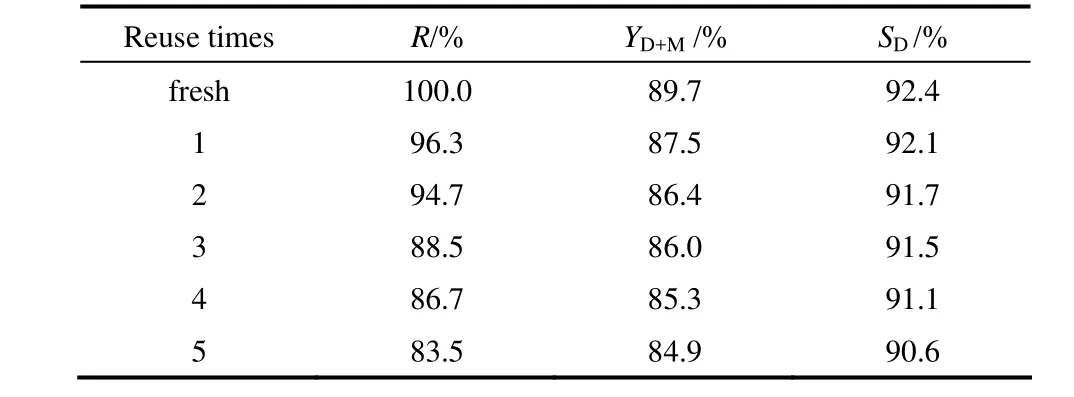
表5 [MIMPS]3PMo12O40的重復性Table 5 Reusability of [MIMPS]3PMo12O40
3 結 論
(1)磺酸功能化離子液體雜多酸鹽中雜多陰離子使其具有較高的熔點,磺酸功能化基團(PS)使其具有很高的催化活性和較好的溶解性。通過實驗證明其兼備均相催化劑與多相催化劑的優點,且用量很少,應用于丁二酸銨水溶液的直接催化酯化表現出良好的催化性能。
(2)功能化有機陽離子(XPS)相同時,陰離子活性順序為[XPS]3PMo12O40> [XPS]4SiW12O40>[XPS]3PW12O40;雜多酸陰離子(Y)相同時,陽離子活性順序為[MIMPS]3Y > [PYPS]3Y > [TEAPS]3Y,但陰陽離子本身對催化劑活性的影響較小,磺酸基團是影響催化劑活性的主要因素。其中[MIMPS]3PMo12O40、[PYPS]3PMo12O40和[TEAPS]3PMo12O40具有較好的催化性能,為本反應理想催化劑。
(3)與傳統催化劑相比,[MIMPS]3PMo12O40具有更好的催化性能,以其作催化劑,得到本反應最佳工藝條件為:催化劑用量1.0%(以丁二酸銨質量計),醇銨摩爾比5:1,丁二酸銨水溶液濃度50%,反應溫度130℃,反應時間12 h,真空度0.02 MPa。該條件下酯收率可達89.7%。
(4)功能化離子液體雜多酸鹽催化劑經多次重復使用后,其結構不變,酯收率仍達85%以上,具有很好的重復使用性能。
[1]Bechthold I, Bretz K, Kabasci S, Kopitzky R, Springer A. Succinic acid: a new platform chemical for biobased polymers from renewable resources [J].Chemical Engineering & Technology, 2008, 31(5):647-654.
[2]Xu Bing(徐冰), Ma Jiangfeng(馬江鋒), Liang Liya(梁麗亞), Liu Rongming(劉嶸明), Wang Guangming(王光明), Zhang Min(張敏),Jiang Min(姜岷). Succinic acid production by repeated batch fermentation based on high density culture [J].CIESC Journal(化工學報), 2011, 62(9): 2595-2599.
[3]Cheng K K, Zhao X B, Zeng J, Zhang J A. Biotechnological production of succinic acid: current state and perspectives [J].Biofuels Bioproducts & Biorefining-Biofpr, 2012, 6(3): 302-318.
[4]Berglund K A, Yedur S, Dunuwila D D. Succinic acid production and purification[P]: US, 5958744.1999.
[5]Li Rongjie(李榮杰), Shang Haitao(尚海濤), Pan Shenglong(潘聲龍),Wu Pengju(吳鵬舉). A method of extracting succinic acid from fermentation broth [P]: CN,101643400SA.2010-2-10.
[6]Berglund K A, Elankovan P, Glassner D A. Carboxylic acid purification and crystallization process[P]: US, 5034105.1991.
[7]Wu Hao(吳昊), Jiang Min(姜岷), Wei Ping(韋萍), Gao Chao(高超),Yang Gao(楊高), Lei Dan(雷丹). Selective separation of succinic acid from simulated fermentation broth by nanofiltration [J].Membrane Science and Technology(膜科學與技術), 2011, 31( 5):46-51.
[8]Kim B S, Hong Y K, Hong W H. Effect of pH on the extraction characteristics of succinic acid and the stability of colloidal liquid aphrons [J].Korean Journal of Chemical Engineering, 2002, 19(4):669-672.
[9]Huh Y S, Jun Y S, Hong Y K, Song H, Lee S Y, Hong W H. Effective purification of succinic acid from fermentation broth produced byMannheimia succiniciproducens[J].Process Biochemistry, 2006,41(6): 1461-1465.
[10]Li Q, Wang D, Wu Y, Li W L, Zhang Y J, Xing J M, Su Z G. One step recovery of succinic acid from fermentation broths by crystallization[J].Separation and Purification Technology. 2010, 72(3): 294-300.
[11]Ye Xiaojin(葉小金), Gong Li(宮莉), Wang Honglei(王紅蕾), Wang Xiaojun(王曉俊), Xu Hongzhang(徐洪章), Xue Donghua(薛冬樺).Extraction of bio-based succinic acid by esterification method [J].Chemistry & Bioengineering(化學與生物工程), 2014, 31(4): 32-36.
[12]Suna X H, Wang Q H, Zhao W C, Ma H Z, Sakata K. Extraction and purification of lactic acid from fermentation broth by esterification and hydrolysis method [J].Separation and Purification Technology,2006, 49(1): 43-48.
[13]Li Weixing(李衛星), Xing Weihong(邢衛紅). Advances in refinement of lactic acid from fermentation broths [J].Chemical Industry and Engineering Progress(化工進展), 2009, 28(3): 491-495.
[14]Zhao W J, Sun X H, Wang Q H, Ma H Z, Teng Y. Lactic acid recovery from fermentation broth of kitchen garbage by esterification and hydrolysis method [J].Biomass & Bioenergy, 2009, 33(1): 21-25.
[15]Budarin V, Luque R, Macquarrie D J, Clark J H. Towards a bio-based industry: benign catalytic esterifications of succinic [J].Chemistry - A European Journal, 2007, 13(24): 6914-6919.
[16]Delhomme C, Goh S L M, Kuhn F E, Weuster-Botz D. Esterification of bio-based succinic acid in biphasic systems: comparison of chemical and biological catalysts [J].J. Mol. Catal. B-Enzymatic,2012, 80: 39-47.
[17]Zhang Yingwei(張英偉), Liu Kang(劉康), Zhang Xinzhi(張新志),Sun Changjiang(孫長江). A method of producing succinic acid [P]:CN, 102807486A. 2012-12-05.
[18]Shi Feng(石峰), Deng Youquan(鄧友全), Peng Jiajian(彭家鍵), Qiao Kun(喬琨), Miao Shaojun(繆少軍). Ionic liquid—a new and clean method for catalytic esterifications [J].Chemistry Online(化學通報),2000, (1).
[19]Li Y, Hu S L, Cheng J H, Lou W Y. Acidic ionic liquid catalyzed esterification of oleic acid for biodiesel synthesis [J].Chinese Journal of Catalysis, 2014, 35(3): 396-406 .
[20]Zhao Dishun(趙地順), Liu Mengshuai(劉猛帥), Xu Zhice(徐智策),Zhang Juan(張娟), Zhang Di(張笛), Fu Jiangtao(付江濤), Ren Peibing(任培兵). Synthesis of dimethyl succinate catalyzed by ionic liquids [J].CIESC Journal(化工學報), 2012, 63(4): 1089-1094.
[21]Zhao Dishun(趙地順), Ge Jingjing(葛京京), Zhai Jianhua(翟建華),Zhang Juan(張娟), Liu Mengshuai(劉猛帥), Li Junpan(李俊盼).Synthesis of succinic acid diisopropyl ester catalyzed by Lewis acid ionic liquids [J].CIESC Journal(化工學報), 2014, 65(2): 561-569.
[22]Leng Y, Wang J, Zhu D R, Ren X Q, Ge H Q, Shen L.Heteropolyanion-based ionic liquids: reaction-induced self-separation catalysts for esterification [J].Angewandte Chemie InternationalEdition, 2009, 48(1): 168-171.
[23]Leng Y, Wang J, Zhu D R, Wu Y J, Zhao P P. Sulfonated organic heteropolyacid salts:recyclable green solid catalysts for esterifications[J].Journal of Molecular Catalysis A:Chemical, 2009, 313(1/2): 1-6.
[24]Wu Q, Dong B Q, Han M H, Zuo Y Z, Jin Y. Synthesis and characterization of novel Bronsted acidic ionic liquids [J].Spectroscopy and Spectral Analysis, 2007, 27(10): 2027-2031.
[25]An Y, Lu L, Li C M, Cheng S F, Gao G H. Oxidative desulfurization catalyzed by molybdophosphate-based ionic liquid [J].Chinese Journal of Catalysis, 2009, 30(12): 1222-1226.
[26]Zhen Bin(甄彬), Li Hansheng(黎漢生), Li Yuan(李原), Wu Qin(吳芹).Catalytic performance of sulfonic acid functionalized ionic liquids on preparation of biodiesel [J].CIESC Journal(化工學報), 2011,62(S2): 80-84.
[27]Harmer M A, Farneth W E, Sun Qun. High surface area Nafion resin silica nanocomposites: a new class of solid acid catalyst [J].Journal of America Chemistry Society, 1996, 118(33): 7708-7715.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
