超臨界流體技術(shù)構(gòu)建殼聚糖納米粒/PLLA-PEG-PLLA復(fù)合微粒及其表征
2015-08-21 06:59:24陳愛(ài)政康永強(qiáng)王士斌唐娜趙暉
化工學(xué)報(bào) 2015年4期
陳愛(ài)政,康永強(qiáng),王士斌,唐娜,趙暉
(1 華僑大學(xué)化工學(xué)院,福建 廈門 361021;2 華僑大學(xué)生物材料與組織工程研究所,福建 廈門 361021)
引 言
核殼結(jié)構(gòu)復(fù)合材料一般由核心與外殼通過(guò)物理或化學(xué)作用形成,其形式一般包括無(wú)機(jī)/有機(jī)、無(wú)機(jī)/無(wú)機(jī)[1]、有機(jī)/有機(jī)[2]以及核心為液體或無(wú)核心的微膠囊和微球。核殼結(jié)構(gòu)復(fù)合材料不僅具有核心和外殼的性能,還因?yàn)楹藲げ牧闲再|(zhì)的相互補(bǔ)充而具有核殼單一材料所不具有的新性能[3],已成為復(fù)合材料、納米材料等領(lǐng)域的研究熱點(diǎn)[4]。
在藥物載體方面,核殼結(jié)構(gòu)復(fù)合材料能夠提高載藥量和包封率、優(yōu)化載體性能、利于包載蛋白質(zhì)或基因和共載藥物等[5]。本課題組在該方面進(jìn)行了較多的探索和研究,Chen 等[6]建立了可用于制備核殼結(jié)構(gòu)的SpEDS 技術(shù),即將納米粒懸浮在殼材的有機(jī)溶液中,噴入高壓釜,與超臨界二氧化碳相互作用,使得殼材在納米粒表面析出,實(shí)驗(yàn)發(fā)現(xiàn)制得的載藥核殼結(jié)構(gòu)復(fù)合材料與非核殼結(jié)構(gòu)載藥載體相比載藥量、包封率和緩釋性能都得到了改善。近年來(lái),核殼結(jié)構(gòu)復(fù)合材料在共載藥物中也得到了廣泛應(yīng)用。共載藥物,特別是共載基因和藥物,在癌癥治療領(lǐng)域中能夠取得良好的聯(lián)合治療效果,有望成為治療癌癥方案中的最佳選擇,因此備受關(guān)注。
目前應(yīng)用于共載載體的復(fù)合材料主要基于無(wú)機(jī)物質(zhì)、脂質(zhì)、聚合物等[7]。無(wú)機(jī)物質(zhì)方面,以二氧化硅的研究較多,但其具有一定的毒性[8]。脂質(zhì)方面,以陽(yáng)離子脂質(zhì)體的研究較多[9],但脂質(zhì)體本身不易儲(chǔ)存,而且應(yīng)用于共載基因和藥物時(shí)其內(nèi)部載藥、外部載基因的結(jié)構(gòu)導(dǎo)致其血液穩(wěn)定性差、網(wǎng)狀內(nèi)皮系統(tǒng)吸附、內(nèi)涵體逃脫阻礙等問(wèn)題,對(duì)基因的保護(hù)和防降解作用不夠[10]。在基于聚合物方面,殼聚糖因具有免疫原性小、無(wú)毒、轉(zhuǎn)染率高及方便修飾的特點(diǎn),廣泛應(yīng)用于藥物、DNA 和RNA[11]的運(yùn)輸。Xu 等[12]用精密造粒技術(shù)(PPF)制備了共載p53 與阿霉素(DOX)的殼聚糖納米粒的雙壁微球,在癌癥治療方面可能具有協(xié)同作用。Liu 等[13]制備了共載卡培他濱和球蛋白的殼聚糖/海藻酸鹽微包納體系,能夠明顯地控制藥物的釋放,而且所載藥物具有順序釋放的性質(zhì)。此外,聚乳酸(polylactic acid,PLLA)是一類生物可降解的醫(yī)用高分子材料,具有良好的生物相容性、藥物緩釋效果和無(wú)免疫原性等特性,已通過(guò)美國(guó)FDA 認(rèn)證,在醫(yī)藥領(lǐng)域中應(yīng)用廣泛[14]。其中聚乳酸衍生物PLLA-PEG-PLLA和聚乳酸-羥基乙酸共聚物(PLGA)具有雙親性,降解速率快,已廣泛應(yīng)用作為基因和藥物載體[15-16]。
核殼結(jié)構(gòu)復(fù)合材料的制備方法有乳液聚合、界面聚合、分散聚合、自組裝和層層沉積等,或是幾種方法的結(jié)合[17],但總體上存在易于聚集、融合、分離和純化產(chǎn)品困難等問(wèn)題。一方面,除去載藥載體中殘留的溶劑和多余藥物需要大量時(shí)間和成本,而且制備過(guò)程使用有機(jī)溶劑,易造成溶劑殘留,會(huì)產(chǎn)生一定的毒性。另一方面,制備過(guò)程條件苛刻,所使用的溫度較高或pH 變化較大,會(huì)導(dǎo)致藥物失活,不適合用于多肽、蛋白質(zhì)類大分子藥物和其他熱敏性物質(zhì)[18-19]。離子凝膠法操作簡(jiǎn)單,不使用有機(jī)溶劑,條件溫和,同時(shí)超臨界流體(SCF)的臨界點(diǎn)(CO2,PC=7.38 MPa,TC=304.1 K)易實(shí)現(xiàn),溶解和擴(kuò)散性能良好,使其擁有溫和的制備條件,適用于熱敏性物質(zhì)的加工[20-21],而且制得的載體有機(jī)溶劑殘留量低,顯示出良好的生物安全性[22],成為藥物劑型制備的熱門方法[23-25]。
基于以上分析,本研究結(jié)合離子凝膠法和SCF技術(shù)各自的優(yōu)點(diǎn),采用SpEDS 技術(shù)制備具有核殼型結(jié)構(gòu)的CS NPs/PLLA-PEG-PLLA 復(fù)合微粒。首先對(duì)CS NPs 的制備條件進(jìn)行考察,其次對(duì)復(fù)合微粒的制備條件進(jìn)行優(yōu)化,最后對(duì)CS NPs 和復(fù)合微粒的理化性質(zhì)和生物相容性進(jìn)行研究,為復(fù)合微粒的載藥藥效考察和實(shí)際應(yīng)用提供理論依據(jù)。
1 實(shí)驗(yàn)材料與方法
1.1 材料與儀器
聚乳酸-聚乙二醇,50×103,濟(jì)南岱罡生物工程有限公司;殼聚糖(200~400 mPa·s,脫乙酰度≥95%)、三聚磷酸鈉(AR,純度≥98%),阿拉丁;磷鎢酸、氫氧化鈉、乙酸、二氯甲烷(AR,純度≥99.5%)、丙酮(AR,純度≥98%)、溴化鉀,國(guó)藥集團(tuán);高糖DMEM 培養(yǎng)基、胎牛血清FBS,美國(guó)Gibico 公司;Alamar Blue,美國(guó)Invitrogen 公司;人體乳腺癌細(xì)胞Bcap-37,中國(guó)科學(xué)院上海生命科學(xué)研究院細(xì)胞資源中心;其余試劑均為進(jìn)口或國(guó)產(chǎn)分析純。
超臨界造粒裝置(同軸噴嘴內(nèi)徑100 μm、外徑500 μm),南通華安超臨界有限公司;掃描電子顯微鏡S-4800、透射電子顯微鏡H-7650,日本Hitachi公司;zeta 電位粒度儀ZetaPALS,英國(guó)馬爾文儀器公司;傅里葉變換紅外光譜儀FTIR 8400S,日本Shimadzu 公司;差示掃描量熱儀DSC 200F3,德國(guó)Netzsch 公司;熱重分析儀DTG-60H,日本島津公司;多功能酶標(biāo)儀SpectraMax M5,美國(guó)Molecular Devices 公司;pH 計(jì)HI8424C,北京哈納科儀器科技有限公司;倒置顯微鏡IX51、數(shù)碼相機(jī)C-5060,日本奧林巴斯光學(xué)工業(yè)株式會(huì)社。
1.2 殼聚糖納米粒的制備
將定量的殼聚糖加到1%乙酸溶液中,充分?jǐn)嚢枞芙猓瑢⑷哿姿徕c加入純化水中,攪拌溶解,分別0.22 μm 微濾后,得到殼聚糖溶液和三聚磷酸鈉溶液。量取一定體積的殼聚糖溶液于試管中,置于渦旋儀上,緩慢加入三聚磷酸鈉溶液,繼續(xù)渦旋1 min,室溫孵育30 min,即可得到CS NPs 溶液。
1.3 復(fù)合微粒的制備
SpEDS 過(guò)程如圖1所示,稱取一定量的PLLA-PEG-PLLA 溶于二氯甲烷中,然后加入一定體積的丙酮作為非溶劑,再加入一定體積的CS NPs溶液,得到不同非溶劑/溶劑比的混合溶液。鋼瓶中的CO2經(jīng)過(guò)制冷系統(tǒng)液化后,由高壓柱塞泵進(jìn)行加壓,泵入結(jié)晶釜中,待釜內(nèi)達(dá)到所要求的壓力時(shí)(12 MPa),維持CO2泵入速率,開啟放氣閥,然后以一定速率放氣(1500 L·h-1),并調(diào)節(jié)結(jié)晶釜外部干燥箱,保持釜內(nèi)壓力、溫度的恒定。當(dāng)達(dá)到實(shí)驗(yàn)所需溫度(35℃)后,ScCO2通過(guò)結(jié)晶釜頂部的同軸噴嘴外側(cè)通道,混合溶液由高壓恒流泵通過(guò)噴嘴內(nèi)側(cè)通道,同時(shí)泵入結(jié)晶釜。結(jié)束泵樣,維持 釜內(nèi)的壓力及溫度不變,繼續(xù)通入新鮮的CO2淋洗20 min。緩慢卸壓,待釜內(nèi)壓力降為常壓時(shí),收集樣品。
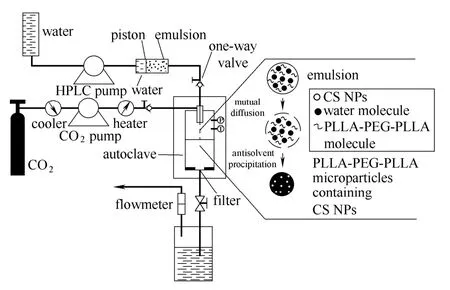
圖1 超臨界抗溶劑制備復(fù)合微粒的過(guò)程Fig.1 Schematic diagram for preparation of MPs by SpEDS
1.4 形貌觀察
(1)粒度檢測(cè):量取一定體積的樣品溶液,采用zeta 電位及納米/亞微米粒度分析儀測(cè)量粒徑和zeta 電位,置于測(cè)量杯中,放入儀器中,用DTS 5.00軟件分析數(shù)據(jù)。
(2)場(chǎng)發(fā)射掃描電子顯微鏡(FE-SEM)觀察:樣品在真空干燥箱中充分干燥,將其置于導(dǎo)電膠上,噴金3 min,置于FE-SEM 下,觀察樣品的表面形態(tài)。
(3)TEM 觀察:配制一定濃度的樣品混懸液,用移液槍吸取10 μl 混懸液滴在銅網(wǎng)上,靜置5 min。用濾紙吸取多余的液體,再向銅網(wǎng)滴加2.0%、pH 7.0的磷鎢酸負(fù)染10 min(僅CS NPs 進(jìn)行負(fù)染),置于暗處充分干燥后,采用TEM 觀察樣品形貌。
1.5 理化表征
(1)傅里葉變換紅外光譜分析(FTIR):取適量KBr 在瑪瑙研缽中研磨成細(xì)粉,然后壓片至透明度較好時(shí),將其作為空白樣,掃描基線。另取適量KBr,研磨成微細(xì)粉末,加入樣品,繼續(xù)研磨,壓片后在波數(shù)4000~400 cm-1范圍內(nèi)檢測(cè)制備前后樣品的特征官能團(tuán)。
(2)差示掃描量熱分析(DSC):取10 mg 左右的樣品,置于Al2O3坩堝中,溫度范圍20~200℃,升溫速率10℃·min-1,載氣氬氣,氣體流量20 μl·min-1。
(3)熱重分析(TGA):在N2流量為50 mg·ml-1的氣氛下,設(shè)定壓力為0.1~0.2 MPa,在20~800℃范圍內(nèi)以10℃·min-1的升溫速率進(jìn)行掃描。
1.6 載體細(xì)胞毒性的考察
(1)取對(duì)數(shù)生長(zhǎng)期的Bcap-37 細(xì)胞,以每孔2×104個(gè)細(xì)胞的密度接種于96 孔培養(yǎng)板上,每孔100 μl,37℃,5% CO2飽和濕度的培養(yǎng)箱中培養(yǎng)。
(2)待細(xì)胞的匯合度約80%時(shí),棄去原液。向各受試組分別加入不同體積的CS NPs 溶液,補(bǔ)齊至每孔200 μl,考察CS NPs 的細(xì)胞毒性;向復(fù)合微粒組的各受試組分別加入培養(yǎng)基超聲分散(200 W,10 min)的復(fù)合微粒溶液(250、500 和 1000 μg·ml-1),每孔200 μl,考察復(fù)合微粒的細(xì)胞毒性。其中,陰性對(duì)照組均加入新鮮培養(yǎng)基。
(3)于加樣后不同時(shí)間點(diǎn)棄去孔板中的培養(yǎng)液,用PBS 緩沖液洗滌2 次,向每孔中加入含有10% Alamar Blue 的新鮮培養(yǎng)基100 μl,在培養(yǎng)箱中繼續(xù)培養(yǎng)4 h。用酶標(biāo)儀檢測(cè)各孔的吸光度值(檢測(cè)波長(zhǎng)570 nm,參考波長(zhǎng)600 nm)。通過(guò)式(1)計(jì)算細(xì)胞的相對(duì)增殖率(relative growth rate,RGR)
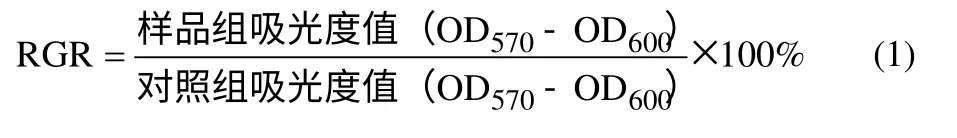
2 結(jié)果與討論
2.1 CS NPs 形貌觀察
經(jīng)過(guò)條件摸索,確定制備CS NPs 的優(yōu)化條件為殼聚糖濃度2 mg·ml-1、pH 5.0、三聚磷酸鈉濃度1 mg·ml-1。從透射電鏡圖2(a)和掃描電鏡圖2(b)可以看出優(yōu)化條件下制備得到的CS NPs 平均粒徑為50~100 nm。該粒徑與粒度分析儀所測(cè)結(jié)果相比,可以發(fā)現(xiàn)粒度分析儀測(cè)得的粒徑是電鏡測(cè)得的3~6 倍。這是因?yàn)镃S 溶解在乙酸溶液中,與TPP 溶液發(fā)生作用后形成的CS NPs 處于膨脹的狀態(tài),而且具有較大的水化層。當(dāng)電鏡觀察時(shí),樣品經(jīng)過(guò)干燥,水分蒸發(fā),發(fā)生了一定程度的皺縮,從而導(dǎo)致兩種測(cè)試結(jié)果不一致。
楊心督等[26]對(duì)聚乙二醇單甲醚接枝殼聚糖自聚集的納米球進(jìn)行粒度表征時(shí)同樣出現(xiàn)該現(xiàn)象,對(duì)此給出的解釋是測(cè)試時(shí)樣品的狀態(tài)導(dǎo)致結(jié)果不一樣。但是兩種測(cè)試方法的粒徑結(jié)果間只相差1 倍,小于本研究存在的差異。這有可能是聚乙二醇單甲醚接枝殼聚糖是一種兩親性分子,具有較小的水化層,而本研究的殼聚糖的親水性較大,因此CS NPs具有較大的水化層,導(dǎo)致倍數(shù)相差更大。
2.2 復(fù)合微粒的制備優(yōu)化
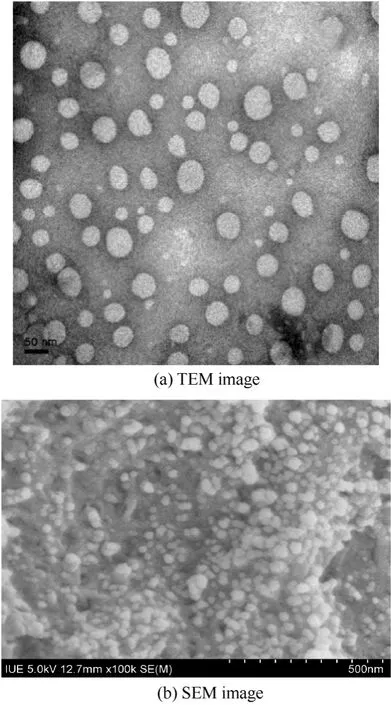
圖2 殼聚糖納米粒的形貌Fig.2 Morphology of CS NPs
2.2.1 全因子設(shè)計(jì)實(shí)驗(yàn) 本實(shí)驗(yàn)中確定的關(guān)鍵變量如下:油相濃度、水油比、溶液流速和溶劑/非溶劑比。首先,濃度對(duì)顆粒粒徑的影響取決于抗溶劑過(guò)程中晶體生長(zhǎng)時(shí)間和生長(zhǎng)速度的平衡關(guān)系,濃度通 過(guò)影響溶液的過(guò)飽和度和黏度最終影響顆粒的粒徑。其次,水油比影響溶液的穩(wěn)定性和抗溶劑效果,而溶液流速的大小除了影響抗溶劑效果外還影響溶液在離開噴嘴后的霧化效果。最后,由于PLLA-PEG-PLLA 溶于二氯甲烷(dichloromethane,DCM)而不溶于丙酮(acetone),丙酮作為非溶劑能夠增大溶液的過(guò)飽和度,從而影響顆粒的粒徑。同時(shí)丙酮的加入有助于CS NPs 的分散,能夠較好地形成包埋CS NPs 的復(fù)合微粒。通過(guò)Minitab 全因子實(shí)驗(yàn)設(shè)計(jì)軟件,考察表1所列的4 個(gè)工藝參數(shù)對(duì)復(fù)合微粒形貌和粒度分布的影響,設(shè)計(jì)出24因子實(shí)驗(yàn)表(表1),并用zeta 電位及納米/亞微米粒度分析儀測(cè)試各實(shí)驗(yàn)組樣品的粒徑和zeta 電位,結(jié)果見(jiàn)表2(每組重復(fù)兩次)。并通過(guò)統(tǒng)計(jì)性分析確定各變量對(duì)復(fù)合微粒粒徑和zeta 電位的顯著性影響及趨勢(shì),進(jìn)而通過(guò)驗(yàn)證實(shí)驗(yàn)得出復(fù)合微粒最佳的制備工藝。
從圖3(a)平均粒徑主效應(yīng)圖可以看出,復(fù)合微粒粒徑隨水油比和溶液流速增大而增大,隨油相濃度增大而減小,但其斜率都較小,表明在所考察的范圍內(nèi)水油比、溶液流速和油相濃度對(duì)復(fù)合微粒最終粒徑的影響不明顯。從圖3(b)平均粒徑效應(yīng)正態(tài)圖可以看出,溶劑/非溶劑比遠(yuǎn)離直線,表明其為影響復(fù)合微粒粒徑的顯著的影響因素。在不改變其他條件的情況下減小溶劑/非溶劑比,可以增大溶液的過(guò)飽和度,使得PLLA-PEG-PLLA 析出更快,有利于形成包覆CS NPs 均一且較小的復(fù)合微粒。zeta 電位方面,從圖3(c)、(d)可以看出,除了水油比外,其他3 個(gè)因素對(duì)復(fù)合微粒的zeta 電位均存在不同程度的影響,但均不顯著。
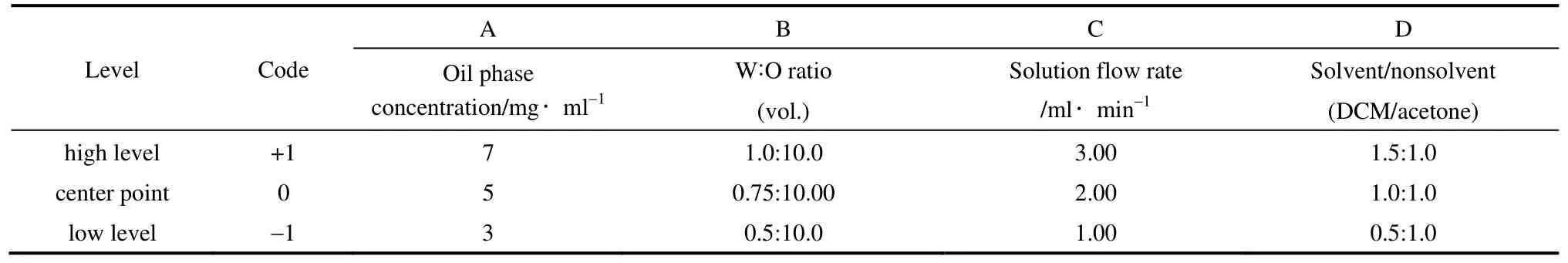
表1 實(shí)驗(yàn)因子和實(shí)驗(yàn)水平Table 1 Experimental factors and levels
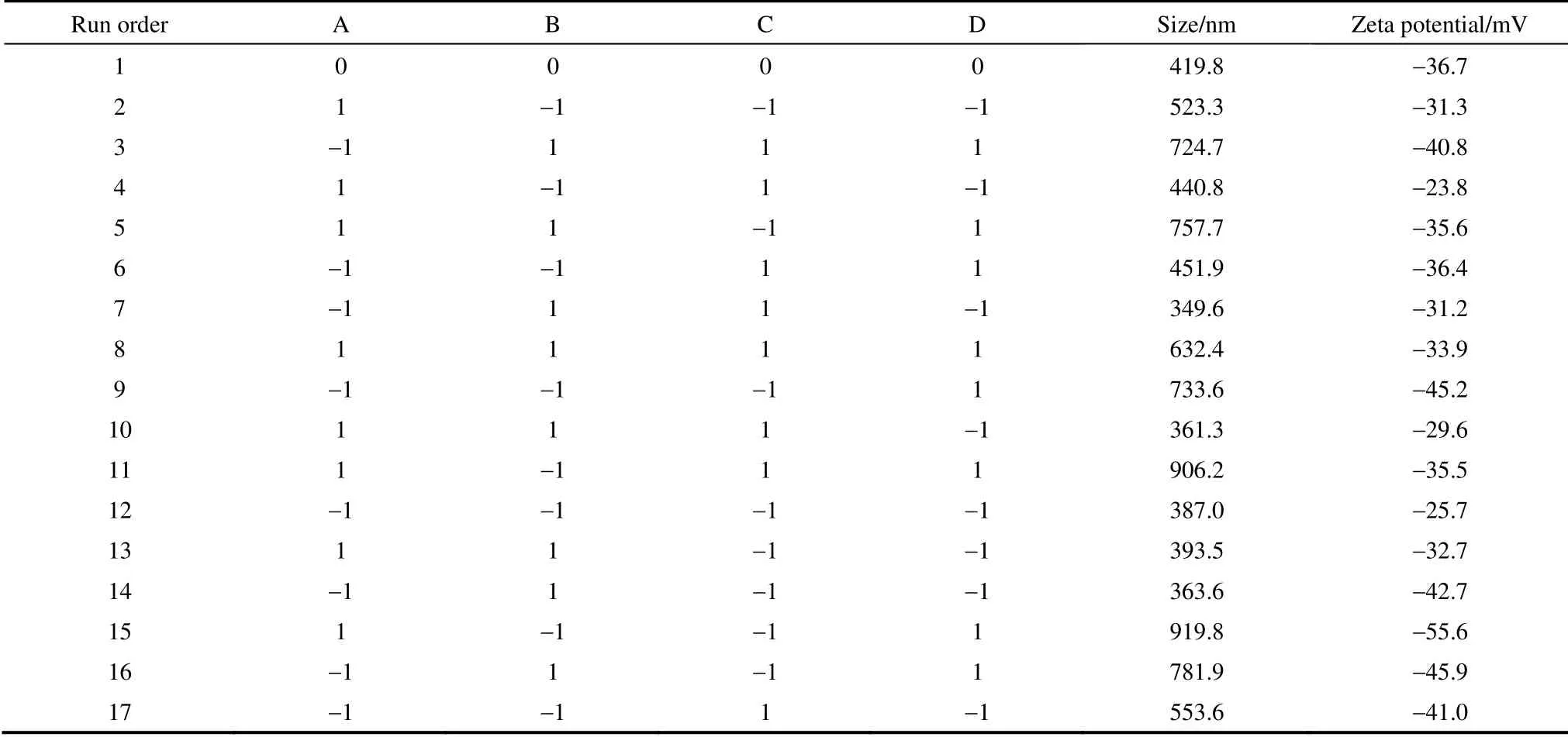
表2 超臨界流體強(qiáng)制分散懸浮液過(guò)程制得復(fù)合微粒的平均粒徑和zeta 電位Table 2 Mean size and zeta potential of MPs prepared by SpEDS
經(jīng)過(guò)進(jìn)一步的分析得到了平均粒徑和zeta 電位的重疊等值線圖(圖4),從而預(yù)測(cè)平均粒徑和zeta 電位符合一定要求(200~400 nm,-50~-30 mV)的復(fù)合微粒制備條件。考慮到水相對(duì)CS NPs 在有機(jī)溶劑中分散效果的影響、樣品收集和載藥藥效的發(fā)揮,最終確定復(fù)合微粒制備的優(yōu)化條件:油相濃度為 5 mg·ml-1、水油比為0.75:10.00、溶液流速為2 ml·min-1、溶劑/非溶劑比為0.5:1.0。
2.2.2 復(fù)合微粒的表面形態(tài) 從圖5可以看出,實(shí)驗(yàn)所得的復(fù)合微粒表面光滑,多為球形。當(dāng)溶劑/非溶劑比為0.5:1.0 時(shí),即Run 2、4、7、10、12、13、14、17 的復(fù)合微粒平均粒徑比其他組更小,說(shuō)明溶劑/非溶劑比是超臨界抗溶劑過(guò)程制備復(fù)合微粒的重要影響因素。
關(guān)于溶劑/非溶劑比在超臨界抗溶劑造粒過(guò)程中的影響已有諸多文獻(xiàn)報(bào)道[27-28],對(duì)顆粒粒徑的影響有兩方面:一方面,高濃度使得溶液較快達(dá)到過(guò)飽和,晶體生長(zhǎng)比晶核生成更占主導(dǎo)地位,而且高濃度也導(dǎo)致溶液黏度變大,二者共同作用使制得的顆粒粒徑變大;另一方面,濃度高使溶液的過(guò)飽和度高,成核速率快,能夠形成粒徑較小的顆粒。基于以上兩方面分析,非溶劑的使用能夠很好地解決上述看似矛盾的現(xiàn)象。這是因?yàn)榉侨軇┑募尤胧谷芤耗軌蚓哂休^低的濃度和較高的過(guò)飽和度,其中低濃度使顆粒形成過(guò)程中成核占主導(dǎo)地位,高過(guò)飽和度使該過(guò)程具有較快的成核速率,從而越有可能獲 得球形度良好、平均粒徑更小的納米粒。本研究通過(guò)增加有機(jī)非溶劑丙酮來(lái)改變材料在溶液中的過(guò)飽和度,有機(jī)非溶劑所占的比例越大,溶液的過(guò)飽和度就越高,在SpEDS 過(guò)程中成核速率就越快,復(fù)合微粒的形成速度也越快,球形度更好,粒度更加均一。
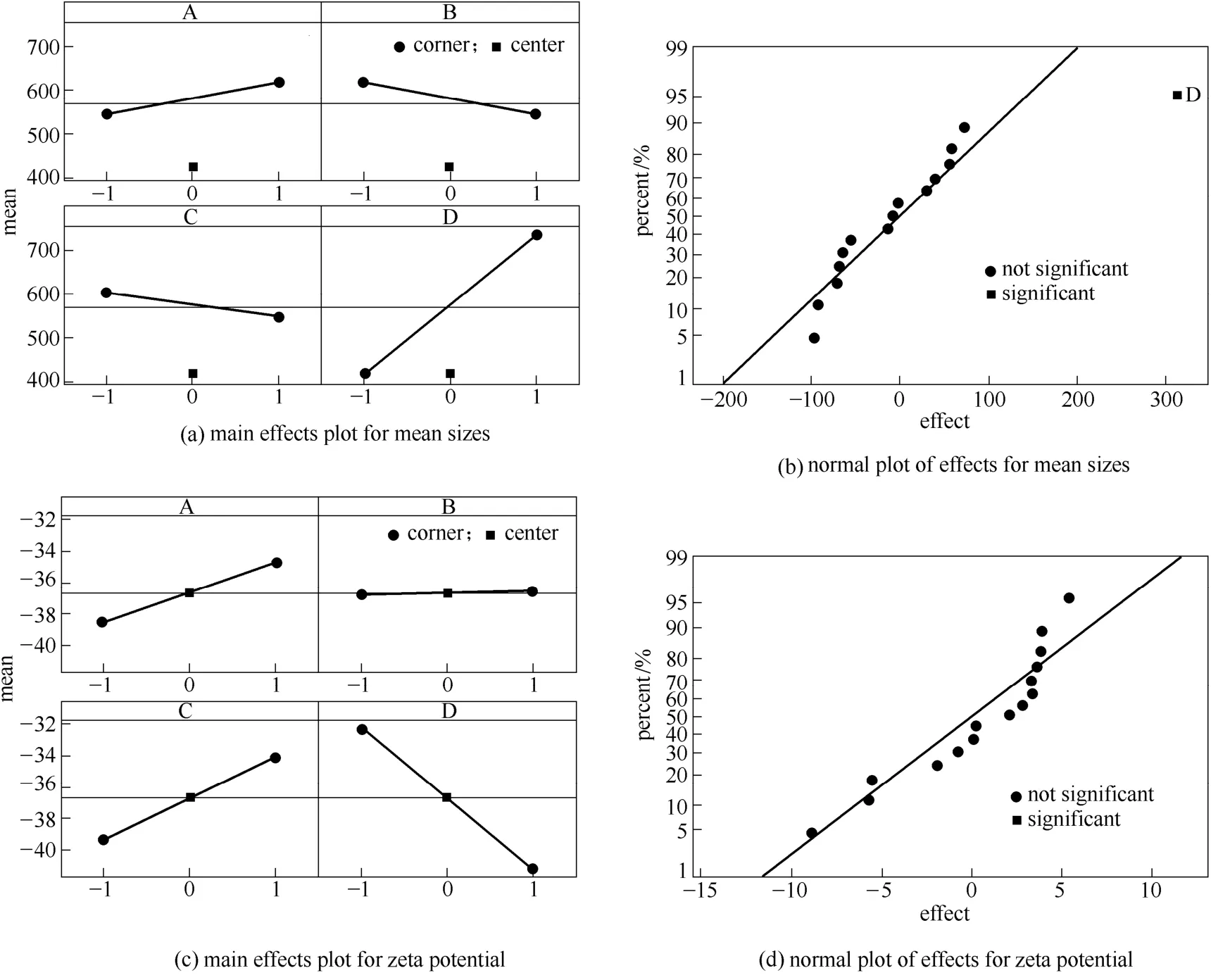
圖3 Minitab 分析結(jié)果Fig.3 Minitab analysis
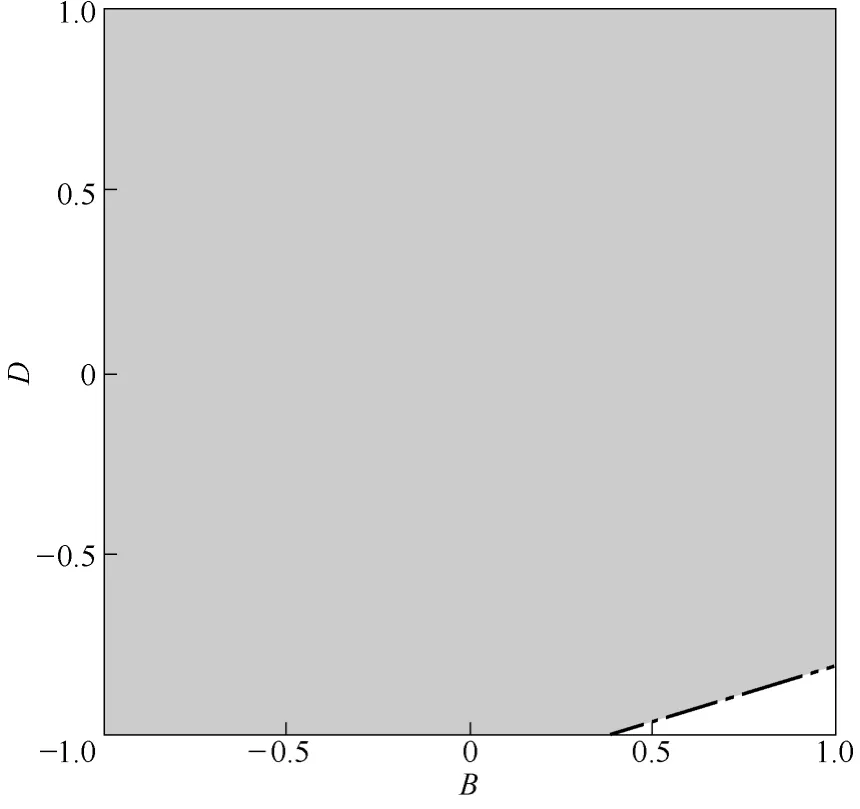
圖4 平均粒徑和zeta 電位的等值線圖Fig.4 Contour plot of mean sizes and zeta potential
2.3 優(yōu)化組復(fù)合微粒的粒度及形貌表征
從圖6可以看出CS NPs 的平均粒徑為314 nm。優(yōu)化組復(fù)合微粒的平均粒徑為323.7 nm,符合Minitab 軟件預(yù)測(cè)的結(jié)果,說(shuō)明該全因子實(shí)驗(yàn)及其預(yù)測(cè)具有可信度。而超純水相優(yōu)化組(MPs without CS NPs)和無(wú)水相優(yōu)化組(MPs without water phase)復(fù)合微粒的平均粒徑分別為412.1 nm 和628.3 nm,說(shuō)明在相同條件下水相和CS NPs 溶液相對(duì)制備得到的微粒粒徑產(chǎn)生了不同的影響。CS NPs 溶液相使制得的復(fù)合微粒粒徑變小,這與之前的研究結(jié)果一樣[6,29]。這是因?yàn)镃S NPs 的存在使得PLLA-PEG- PLLA 在一定程度上析出并附著在CS NPs 表面,而非形成獨(dú)立的PLLA-PEG-PLLA 微粒。同時(shí)CS NPs 起晶核的作用,降低了PLLA-PEG- PLLA 析出的能壘,使其容易快速在CS NPs 表面析出,形成更小的微粒。從圖7復(fù)合微粒透射電鏡圖可以看出,CS NPs 包埋在PLLA-PEG- PLLA 中,形成核殼型的復(fù)合微粒,經(jīng)Nano Measurer 軟件測(cè)量,該復(fù)合微粒的粒徑比圖6粒徑測(cè)試的結(jié)果小,說(shuō)明PLLA-PEG-PLLA 材料的親水性導(dǎo)致復(fù)合微粒表面尚存在一定的水化層。

圖5 不同制備條件下所得復(fù)合微粒的掃描電鏡圖Fig.5 SEM images of MPs obtained in different run orders
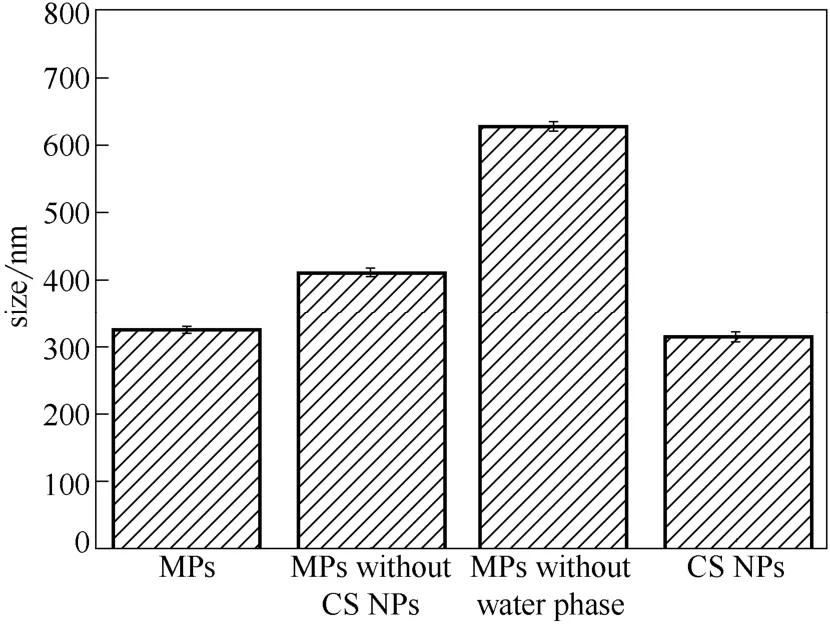
圖6 優(yōu)化組復(fù)合微粒的平均粒徑Fig.6 Mean sizes of MPs under optimal condition
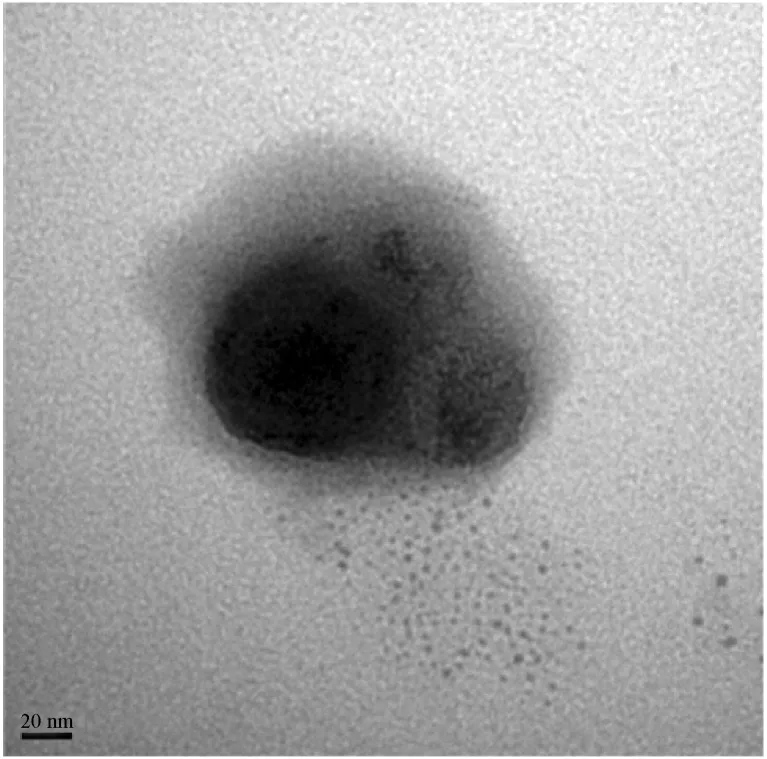
圖7 優(yōu)化組復(fù)合微粒的透射電鏡圖Fig.7 TEM image of MPs under optimal condition
2.4 FTIR 表征
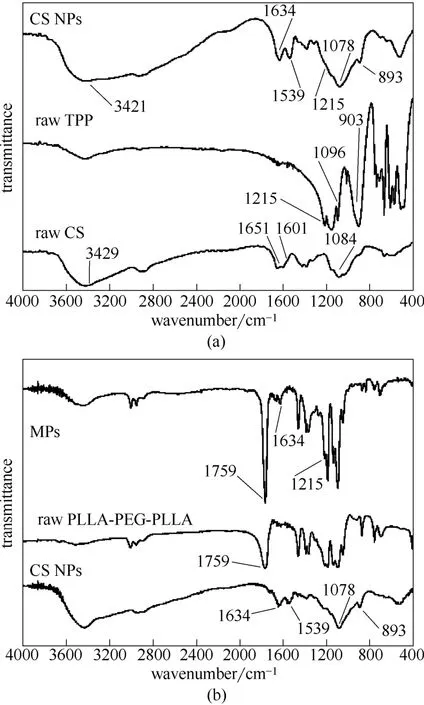
圖8 殼聚糖納米粒和復(fù)合微粒的紅外光譜圖Fig.8 FTIR spectra of CS NPs and MPs
從CS 原材料、TPP 原材料和CS NPs 的紅外光譜[圖8(a)]可以看出:原料CS 方面,3429 cm-1為—OH 的伸縮振動(dòng)吸收峰,1651 cm-1和1601 cm-1分別為酰胺鍵和—NH2的吸收峰,1084 cm-1為C—O—C 的伸縮振動(dòng)吸收峰;TPP 方面,1096 cm-1和903 cm-1分別是TPP 的P—O 鍵伸縮振動(dòng)吸收峰和彎曲振動(dòng)吸收峰,1215 cm-1處為伸縮吸收峰;CS NPs 方面,吸收峰有一定右移,可能是 因?yàn)門PP 與CS 靜電結(jié)合后影響CS 分子的電荷密度,導(dǎo)致其官能團(tuán)吸收峰發(fā)生一定程度的藍(lán)移現(xiàn)象,具體如—OH 的伸縮振動(dòng)吸收峰遷移至3421 cm-1、1651 cm-1和1601 cm-1分別遷移至1634 cm-1和1539 cm-1,同時(shí)CS NPs 中1078 cm-1和893 cm-1分別出現(xiàn)TPP 的P—O 鍵伸縮振動(dòng)吸收峰和彎曲振動(dòng)吸收峰以及在1215 cm-1處有伸縮吸收峰。上述官能團(tuán)吸收峰的變化情況與文獻(xiàn)報(bào)道[30-31]一致。由此可推測(cè),TPP 的磷酸根與CS 的氨基位點(diǎn)作用,形成了CS NPs。如紅外圖譜圖8(b)所示,在優(yōu)化組復(fù)合微粒中1634 cm-1和1215 cm-1均有各自的吸收峰,應(yīng)為CS NPs 中所含有的特征吸收峰,表明 CS NPs 存在復(fù)合微粒中。另外,PLLA-PEG-PLLA 原料和復(fù)合微粒均在1759 cm-1處找到特征吸收峰且無(wú)新的特征峰出現(xiàn),表明該超臨界抗溶劑過(guò)程是一個(gè)物理過(guò)程,不涉及化學(xué)變化。這與Chen 等[32]的研究結(jié)果一致,說(shuō)明超臨界流體過(guò)程只是改變物質(zhì)的物理狀態(tài),不引起官能團(tuán)的明顯變化,適合應(yīng)用于藥物載體的制備。
2.5 TGA 表征
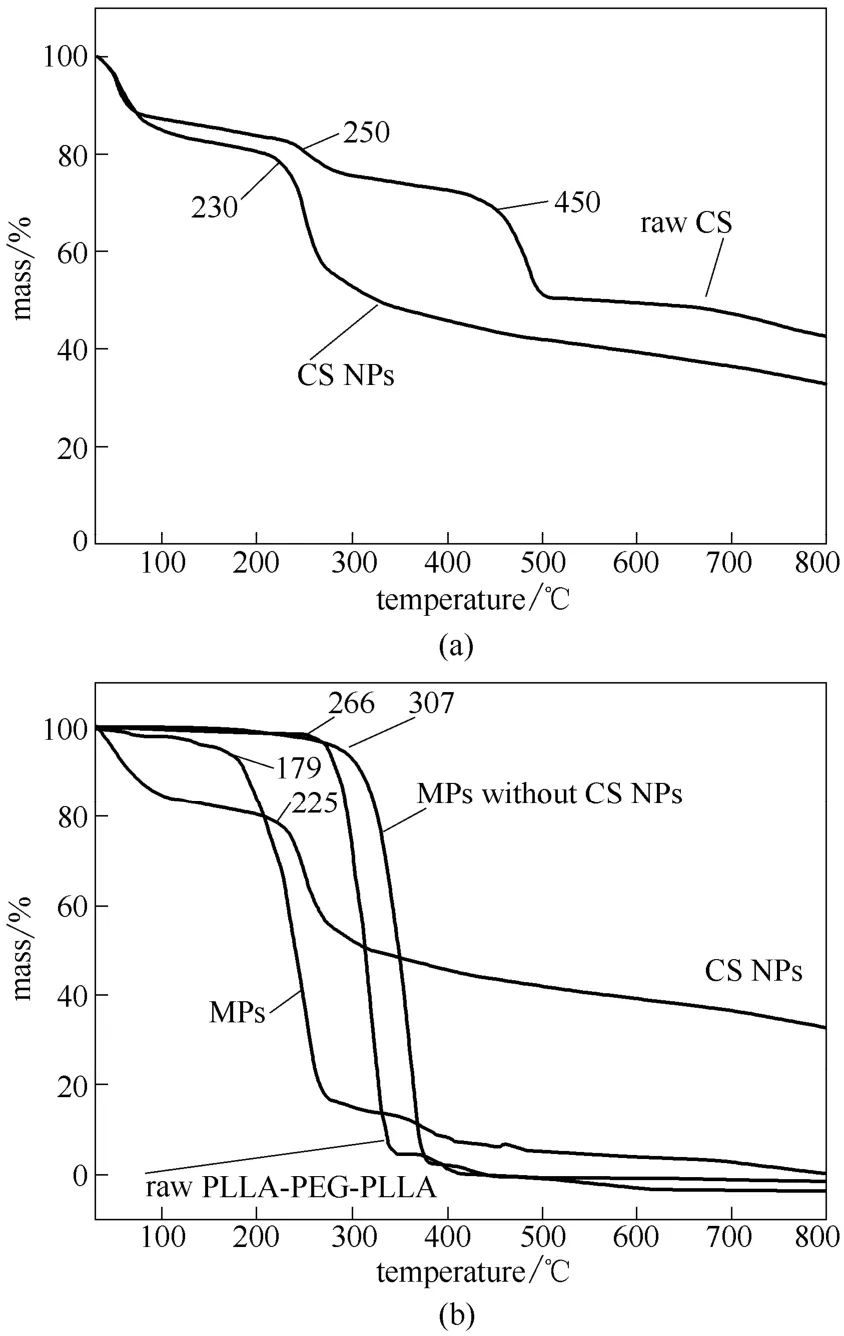
圖9 殼聚糖納米粒和復(fù)合微粒的熱重曲線Fig.9 TGA curves of CS NPs and MPs
如熱重曲線圖9(a)所示,100℃之前原料CS 和CS NPs 都有一定的質(zhì)量損失(15%~20%),是由于樣品中殘留的水分引起的。CS NPs 的分解溫度在230℃附近,而原料CS 的分解溫度開始于250℃,并在450℃附近質(zhì)量急劇下降,該結(jié)果與李國(guó)明等[33]的研究類似。其中CS 和TPP 通過(guò)靜電結(jié)合后氫鍵受到破壞,是其分解溫度有所降低的重要原因。一方面,CS 的氨基與TPP 的磷酸根通過(guò)靜電結(jié)合,氫鍵減少;另一方面,CS NPs 形成過(guò)程中部分基團(tuán)以氫鍵的形式與水結(jié)合,使得CS 分子中以氫鍵連接的連接點(diǎn)減少,這也從另一方面說(shuō)明了CS NPs的形成。如圖9(b)所示,從CS NPs 和優(yōu)化組復(fù)合微粒的熱重圖可以看出,100℃之前CS NPs 有質(zhì)量損失,是由于CS NPs 中水分蒸發(fā)所致。CS NPs從225℃開始出現(xiàn)了質(zhì)量的急劇損失,是由于CS NPs 在此溫度后開始分解所致。復(fù)合微粒從179℃起質(zhì)量急劇下降,而原料PLLA-PEG-PLLA 的分解溫度為266℃。二者分解溫度的差異,可能是因?yàn)閮?yōu)化組復(fù)合微粒所包裹的CS NPs 中的水分蒸發(fā),同時(shí)也引發(fā)經(jīng)超臨界處理后的CS NPs 提前開始分解。此外,超純水相復(fù)合微粒的分解溫度為307℃,高于原料PLLA-PEG-PLLA 的分解溫度,說(shuō)明經(jīng)過(guò)SpEDS 過(guò)程后材料PLLA-PEG-PLLA 的熱穩(wěn)定性能有所提高,同時(shí)也證實(shí)優(yōu)化組復(fù)合微粒分解溫度為179℃是由于其含有CS NPs 造成的。在800℃之前優(yōu)化組復(fù)合微粒所剩下的質(zhì)量分?jǐn)?shù)比超純水相復(fù)合微粒多,說(shuō)明優(yōu)化組復(fù)合微粒中含有CS NPs,這一點(diǎn)可由優(yōu)化組復(fù)合微粒的透射電鏡圖得到驗(yàn)證。
2.6 DSC 表征
如差熱曲線圖10(a)所示,CS NPs 和原料CS 均在60~70℃之間有較寬的吸熱峰,峰值分別為65.6℃和69.1℃,這是水分蒸發(fā)吸熱所致。此外,CS NPs 和原料CS 分別在271.4℃和大于300℃的位置出現(xiàn)了放熱峰,表明物質(zhì)開始分解。

圖10 殼聚糖納米粒和復(fù)合微粒的差熱曲線Fig.10 DSC curves of CS NPs and MPs
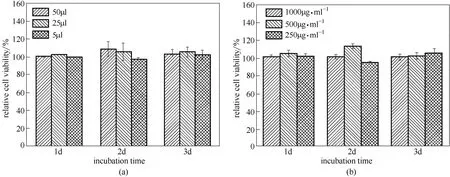
圖11 Alamar Blue 法測(cè)定殼聚糖納米粒及其復(fù)合微粒的細(xì)胞毒性Fig.11 Cytotoxicity of CS NPs and MPs evaluated by Alamar Blue assay
李國(guó)明等[33]報(bào)道制備了載藥殼聚糖微球,其中DSC 結(jié)果顯示原料CS 的分解溫度為310℃,而形成的空白殼聚糖微球分解溫度提前至260℃,與本研究的DSC 結(jié)果一致,說(shuō)明CS 和TPP 通過(guò)靜電結(jié)合后,由于氫鍵被破壞,其分解溫度有所降低,故CS NPs 的分解吸熱峰比原料CS 提前。如圖10(b)所示,原料PLLA-PEG-PLLA 的兩個(gè)熔融吸熱峰分別是170.6℃和177.4℃,說(shuō)明在材料中存在兩種不同結(jié)晶區(qū)[34],而復(fù)合微粒在此處只存在170.6℃單一吸熱峰,說(shuō)明經(jīng)過(guò)ScCO2抗溶劑處理后材料的高溫結(jié)晶區(qū)消失或向低溫結(jié)晶區(qū)轉(zhuǎn)變,晶型更加均勻。此外,優(yōu)化組復(fù)合微粒中131.6℃和170.6℃分別代表所含CS NPs 中的水分蒸發(fā)的吸熱峰以及復(fù)合微粒的熔融吸熱峰。與CS NPs 相比,復(fù)合微粒中的CS NPs 水分蒸發(fā)的吸熱峰從 65.6 ℃移動(dòng)到131.6℃,有可能是因?yàn)镻LLA-PEG-PLLA 材料對(duì)CS NPs 有一定的包封作用并形成核殼型復(fù)合微粒,產(chǎn)生了熱傳遞阻礙作用,導(dǎo)致水分的蒸發(fā)過(guò)程需要更高的溫度才能進(jìn)行。張艷輝等[35]在研究乙二醇雙硬脂酸酯/PMMA 核殼儲(chǔ)能微膠囊時(shí)也發(fā)現(xiàn)經(jīng)包埋后微膠囊的熔點(diǎn)峰變大,說(shuō)明殼材對(duì)核材的傳熱有阻隔作用,從而出現(xiàn)滯后的現(xiàn)象。然而,Kang 等[36]制備包載牛血清蛋白的聚乳酸微粒時(shí)發(fā)現(xiàn)經(jīng)包載后的牛血清蛋白的熔融峰變小,這是因?yàn)槿廴诜宓淖冃∨c牛血清蛋白的濃度低以及自身的螺旋折疊結(jié)構(gòu)有關(guān)。因此,熔融峰的改變與載體材料性質(zhì)和載體結(jié)構(gòu)都有一定的關(guān)系。
2.7 載體的細(xì)胞毒性
從圖11得知,各個(gè)加樣量的實(shí)驗(yàn)組的細(xì)胞相對(duì)增殖率均在100%附近。通過(guò)分級(jí)標(biāo)準(zhǔn)表3可知,CS NPs 和復(fù)合微粒的細(xì)胞毒性處于0 級(jí),安全系數(shù)高。該結(jié)果說(shuō)明實(shí)驗(yàn)中制備得到的CS NPs和復(fù)合微粒的生物相容性好。Chen 等[22]將PLLA-PEG-PLLA 用于包埋具有一定毒性的四氧化三鐵納米粒,能夠明顯降低細(xì)胞毒性,說(shuō)明PLLA-PEG-PLLA 的生物相容性好。本研究中CS也是生物相容性好的高分子材料,導(dǎo)致CS NPs和復(fù)合微粒的生物相容性無(wú)明顯差別,也說(shuō)明本研究所采用的離子凝膠法和超臨界抗溶劑過(guò)程是綠色環(huán)保的載體制備方法,所制得的載體生物相容性良好。

表3 細(xì)胞相對(duì)增殖率分級(jí)標(biāo)準(zhǔn)Table 3 Criterion of cell relative growth rate gradation
3 結(jié) 論
本研究利用離子凝膠法和超臨界強(qiáng)制分散懸浮液技術(shù)分別制得CS NPs 與CS NPs/PLLA-PEG- PLLA 復(fù)合微粒,進(jìn)行了制備條件的考察和優(yōu)化、理化性質(zhì)表征和生物相容性研究等一系列實(shí)驗(yàn)。結(jié)果表明:① 優(yōu)化條件下制得的CS NPs,其表面較光滑,球形度好,粒徑分布窄;② 超臨界強(qiáng)制分散懸浮液技術(shù)過(guò)程中,溶劑/非溶劑對(duì)復(fù)合微粒的粒徑具有顯著影響,優(yōu)化后的復(fù)合微粒具有一定的核殼型結(jié)構(gòu),而且其官能團(tuán)未發(fā)生明顯的變化,但材料的物理形態(tài)發(fā)生了一定的改變;③ CS NPs 及其復(fù)合微粒無(wú)明顯細(xì)胞毒性,具有良好的生物相容性。
以上結(jié)果,將有利于核殼結(jié)構(gòu)的復(fù)合微粒進(jìn)一步應(yīng)用于共載基因和藥物的藥效研究中,有望在癌癥治療領(lǐng)域取得良好的聯(lián)合治療效果。
[1]Li C Y,Ma C,Wang F,Xi Z J,Wang Z F,Deng Y,He N Y.Preparation and biomedical applications of core-shell silica/magnetic nanoparticle composites [J].Journal of Nanoscience and Nanotechnology,2012,12(4):2964-2972
[2]Mandal B,Bhattacharjee H,Mittal N,Sah H,Balabathula P,Thoma L A,Wood G C.Core-shell-type lipid-polymer hybrid nanoparticles as a drug delivery platform [J].Nanomedicine:Nanotechnology,Biology and Medicine,2013,9(4):474-491
[3]Wang Wei (汪偉),Xie Rui (謝銳),Ju Xiaojie (巨曉潔),Chu Liangyin (褚良銀).Recent progress of microfluidic fabrication of novel functional microparticles [J].CIESC Journal(化工學(xué)報(bào)),2014,65(7):2555-2562
[4]Kamata K,Lu Y,Xia Y N.Synthesis and characterization of monodispersed core-shell spherical colloids with movable cores [J].Journal of the American Chemical Society,2003,125(9):2384-2385
[5]Yang Y Y,Wang Y,Powell R,Chan P.Polymeric core-shell nanoparticles for therapeutics [J].Clinical and Experimental Pharmacology and Physiology,2006,33(5/6):557-562
[6]Chen A Z,Li Y,Chau F T,Lau T Y,Hu J Y,Zhao Z,Mok D K W.Microencapsulation of puerarin nanoparticles by poly(L-lactide) in a supercritical CO2process [J].Acta Biomaterialia,2009,5(8):2913-2919
[7]Creixell M,Peppas N A.Co-delivery of siRNA and therapeutic agents using nanocarriers to overcome cancer resistance [J].Nano Today,2012,7(4):367-379
[8]Huang H C,Barua S,Sharma G,Dey S K,Rege K.Inorganic nanoparticles for cancer imaging and therapy [J].Journal of Controlled Release,2011,155(3):344-357
[9]Saad M,Garbuzenko O B,Minko T.Co-delivery of siRNA and an anticancer drug for treatment of multidrug-resistant cancer [J].Nanomedicine,2008,3(6):761-776
[10]Zhang Yingying (張瑩瑩),Chen Jianming (陳建明).Existing problems and strategies in liposome-mediated nucleic acid delivery [J].Pharmaceutica Sinica(藥學(xué)學(xué)報(bào)),2011,46(3):261-268
[11]Lee K Y.Chitosan and its derivatives for gene delivery [J].Macromolecular Research,2007,15(3):195-201
[12]Xu Q X,Xia Y J,Wang C H,Pack D W.Monodisperse double-walled microspheres loaded with chitosan-p53 nanoparticles and doxorubicin for combined gene therapy and chemotherapy [J].Journal of Controlled Release,2012,163(2):130-135
[13]Liu Y G,Sun X Z,Wang S B,Xie M B,Chen A Z,Long R M.Preparation of nanoparticles embedded microcapsules (NEMs) and their application in drug release [J].Materials Letters,2012,75:48-50
[14]Palm M D,Goldman M P.Patient satisfaction and duration of effect with PLLA:a review of the literature [J].Journal of Drugs in Dermatology,2009,8(10):S15-S20
[15]Bala I,Hariharan S,Kumar M.PLGA nanoparticles in drug delivery:the state of the art [J].Critical Reviews in Therapeutic Drug Carrier Systems,2004,21(5):387-422
[16]Wang H J,Zhao P Q,Su W Y,Wang S,Liao Z Y,Niu R F,Chang J.PLGA/polymeric liposome for targeted drug and gene co-delivery [J].Biomaterials,2010,31(33):8741-8748
[17]Cao S S,Chen J R,Hu J.The fabrication and progress of core-shell composite materials [J].Australian Journal of Chemistry,2009,62(12):1561-1576
[18]Rahimi M,Wadajkar A,Subramanian K,Yousef M,Cui W N,Hsieh J T,Nguyen K T.In vitroevaluation of novel polymer-coated magnetic nanoparticles for controlled drug delivery [J].Nanomedicine:Nanotechnology,Biology and Medicine,2010,6(5):672-680
[19]Sun C,Lee J S H,Zhang M Q.Magnetic nanoparticles in MR imaging and drug delivery [J].Advanced Drug Delivery Reviews,2008,60(11):1252-1265
[20]Prosapio V,Reverchon E,De Marco I.Antisolvent micronization of BSA using supercritical mixtures carbon dioxide plus organic solvent [J].Journal of Supercritical Fluids,2014,94:189-197
[21]Kang Y Q,Zhao C,Chen A Z,Wang S B,Liu Y G,Wu W G,Su X Q.Study of lysozyme-loaded poly-L-lactide (PLLA) porous microparticles in a compressed CO2antisolvent process [J].Materials,2013,6(8):3571-3583
[22]Chen A Z,Li L,Wang S B,Lin X F,Liu Y G,Zhao C,Wang G Y,Zhao Z.Study of Fe3O4-PLLA-PEG-PLLA magnetic microspheres based on supercritical CO2:preparation,physicochemical characterization,and drug loading investigation [J].Journal of Supercritical Fluids,2012,67:139-148
[23]Reverchon E,Adami R,Cardea S,Porta G D.Supercritical fluids processing of polymers for pharmaceutical and medical applications [J].Journal of Supercritical Fluids,2009,47(3):484-492
[24]Deng Aihua (鄧愛(ài)華),Chen Aizheng (陳愛(ài)政),Wang Shibin (王士斌),Wang Mingzong (王明宗).Preparation and characterization of silk fibroin nanoparticles by SEDS process [J].Chemical Industry and Engineering Progress(化工進(jìn)展),2014,33(6):1506-1512
[25]Yu Jian (余堅(jiān)),He Jiasong (何嘉松).Fundamental issues for microfoaming polymers with supercritical CO2technology [J].Scientia Sinica:Chimica(中國(guó)科學(xué):化學(xué)),2010,40(1):1-15
[26]Yang Xindu (楊心督),Chen Han (陳漢),Zhang Huizhu (張惠珠),Gao Fuping (高福平),Wang Yinsong (王銀松),Zhang Qiqing (張其清).Preparation of self-assembled monomethoxy poly(ethylene glycol)-grafted-chitosan nanoparticles [J].Chinese Journal of New Drugs(中國(guó)新藥雜志),2007,16(21):1780-1783,1787
[27]Chen A Z,Li Y,Chau F T,Lau T Y,Hu J Y,Zhao Z,Mok D K W.Application of organic nonsolvent in the process of solution-enhanced dispersion by supercritical CO2to prepare puerarin fine particles [J].Journal of Supercritical Fluids,2009,49(3):394-402
[28]Chen A Z,Li L,Wang S B,Zhao C,Liu Y G,Wang G Y,Zhao Z.Nanonization of methotrexate by solution-enhanced dispersion by supercritical CO2[J].Journal of Supercritical Fluids,2012,67:7-13
[29]Kang Y Q,Yin G F,Ouyang P,Huang Z B,Yao Y D,Liao X M,Chen A Z,Pu X M.Preparation of PLLA/PLGA microparticles using solution enhanced dispersion by supercritical fluids (SEDS) [J].Journal of Colloid and Interface Science,2008,322(1):87-94
[30]Dudhani A R,Kosaraju S L.Bioadhesive chitosan nanoparticles:preparation and characterization [J].Carbohydrate Polymers,2010,81(2):243-251
[31]Xu Y M,Du Y M.Effect of molecular structure of chitosan on protein delivery properties of chitosan nanoparticles [J].International Journal of Pharmaceutics,2003,250(1):215-226
[32]Chen A Z,Wang G Y,Wang S B,Li L,Liu Y G,Zhao C.Formation of methotrexate-PLLA-PEG-PLLA composite microspheres by microencapsulation through a process of suspension-enhanced dispersion by supercritical CO2[J].International Journal of Nanomedicine,2012,7:3013-3022
[33]Li Guoming (李國(guó)明),Ye Junsheng (葉俊生),Deng Guohong (鄭國(guó)紅),Liu Cong (劉聰),Wang Chaoyang (汪朝陽(yáng)).Preparation of levodopa-chitosan microspheres and the drug-release performance [J].Chinese Journal of Applied Chemistry(應(yīng)用化學(xué)),2007,24(9):1602-1605
[34]Narita J,Katagiri M,Tsuji H.Highly enhanced accelerating effect of melt-recrystallized stereocomplex crystallites on poly (L-lactic acid) crystallization,2-effects of poly (D-lactic acid) concentration [J].Macromolecular Materials and Engineering,2013,298(3):270-282
[35]Zhang Yanhui (張艷輝),Deng Jianguo (鄧建國(guó)),Huang Yigang (黃奕剛).Preparation of phase change material EGDS/PMMA core-shell microcapsules [J].Chemical Industry and Engineering Progress(化工進(jìn)展),2012,31(3):580-585
[36]Kang Y Q,Yang C,Ouyang P,Yin G F,Huang Z B,Yao Y D,Liao X M.The preparation of BSA-PLLA microparticles in a batch supercritical anti-solvent process [J].Carbohydrate Polymers,2009,77(2):244-249
猜你喜歡
房地產(chǎn)導(dǎo)刊(2022年5期)2022-06-01 06:20:14
建材發(fā)展導(dǎo)向(2021年12期)2021-07-22 08:06:48
建材發(fā)展導(dǎo)向(2021年7期)2021-07-16 07:07:52
河北科技師范學(xué)院學(xué)報(bào)(2021年1期)2021-05-10 03:34:20
中學(xué)生數(shù)理化(高中版.高二數(shù)學(xué))(2021年12期)2021-04-26 07:43:48
中學(xué)生數(shù)理化(高中版.高考數(shù)學(xué))(2021年12期)2021-03-08 01:28:50
電源技術(shù)(2017年1期)2017-03-20 13:37:59
食品界(2016年4期)2016-02-27 07:36:46
現(xiàn)代企業(yè)(2015年2期)2015-02-28 18:45:09
應(yīng)用化工(2014年7期)2014-08-09 09:20:21
