載體對鎳基催化劑順酐液相加氫性能的影響
2015-08-22 11:09:34張因趙麗麗張鴻喜李海濤劉盼盼蓋媛媛趙永祥
化工學報 2015年7期
關鍵詞:催化劑
張因,趙麗麗,張鴻喜,李海濤,劉盼盼,蓋媛媛,趙永祥
(山西大學精細化學品教育部工程研究中心,山西 太原 030006)
引 言
目前,人們已開展了順酐氣相、液相以及超臨界CO2流體中加氫的研究,涉及的催化劑主要有貴金屬Pt、Pd基以及非貴金屬Cu、Ni基催化劑。貴金屬Pt、Pd催化劑主要應用于順酐液相以及超臨界CO2流體中加氫,加氫產(chǎn)物主要有丁二酸酐、丁酸、γ-丁內(nèi)酯等[2-5];Cu基催化劑主要應用于順酐氣相加氫,丁二酸酐、γ-丁內(nèi)酯、四氫呋喃和1,4-丁二醇是主要產(chǎn)物[6-8];Ni基催化劑多應用于順酐液相及氣相加氫,主要產(chǎn)物是丁二酸酐和γ-丁內(nèi)酯[1,9-17]。在上述催化體系中,Ni基催化劑因其低廉的成本和高的活性、選擇性而引起了研究者極大的關注[9-10,14-17]。本課題組曾系統(tǒng)研究了Ni-SiO2催化劑[9-10,18-21]的順酐液相加氫性能。發(fā)現(xiàn)活性組分Ni物種的存在形態(tài)以及其與載體間的相互作用,對產(chǎn)物選擇性有重要影響。當Ni物種以微細簇團形式存在并與載體有強相互作用時,順酐加氫主要生成γ-丁內(nèi)酯;當Ni以結(jié)晶態(tài)存在并與載體相互作用弱時,順酐加氫的主產(chǎn)物是丁二酸酐[18-21]。進一步的研究發(fā)現(xiàn),在SiO2中引入ZrO2對載體表面性質(zhì)改性后,催化劑的選擇性發(fā)生顯著改變,順酐加氫主產(chǎn)物由丁二酸酐轉(zhuǎn)變?yōu)棣?丁內(nèi)酯[9]。最近,Guo等[15]及Marchi 等[14,22]也關注到了載體對催化劑順酐加氫性能的影響,他們研究了硅藻土、γ-Al2O3、膨潤土、凹凸棒土、SiO2、SiO2-Al2O3、H-BEA沸石負載金屬催化劑的順酐加氫性能。發(fā)現(xiàn)載體可通過影響活性金屬與載體間的相互作用來改變催化劑的加氫性能:當載體與活性組分產(chǎn)生強的相互作用時,活性組分以較高分散度存在,其加氫活性顯著高于金屬-載體相互作用弱的催化劑。這與本課題組前期研究結(jié)論一致。在巴豆醛選擇加氫中,Vannice等[23]研究了不同載體負載Pt催化劑的巴豆醛選擇加氫性能,發(fā)現(xiàn)載體可通過與反應物產(chǎn)生相互作用,來改善催化劑的加氫選擇性。但是,在順酐加氫中,載體是否可與反應物產(chǎn)生吸附作用,而不是僅通過改善活性組分的分散度來影響催化劑的順酐加氫產(chǎn)物選擇性,還不甚明了,也未見相關的文獻報道。
基于此,本工作分別以不同性質(zhì)的ZrO2、SiO2以及Al2O3為載體,研究其負載鎳催化劑的順酐液相加氫性能,并采用套用實驗考察其使用穩(wěn)定性,結(jié)合H2-TPR、XRD、TPO-MS等表征手段,深入理解鎳基催化劑催化順酐加氫中的載體效應。
1 實驗部分
1.1 載體的制備
采用溶膠-凝膠結(jié)合超臨界干燥技術制備ZrO2、SiO2以及Al2O3載體,具體的制備步驟如下:①ZrO2凝膠,稱取一定量硝酸氧鋯[ZrO(NO3)2·2H2O]溶于無水乙醇中,加入醇水體積比為4:1的水,置于333 K的恒溫水浴中緩慢攪拌得ZrO2凝膠;②SiO2凝膠,SiO2凝膠的制備步驟同ZrO2,其中SiO2凝膠制備的反應介質(zhì)中醇水體積比為1:1;③Al2O3凝膠,稱取一定量硝酸鋁[Al(NO3)3·9H2O]溶于無水乙醇中,加入與Al(NO3)3·9H2O摩爾比為3:1的尿素,與醇水體積比為1:1.6的水,混合均勻后置于368 K的恒溫油浴中回流得到Al2O3凝膠。將上述凝膠經(jīng)乙醇超臨界干燥后置于馬弗爐中773 K焙燒3 h備用。
1.2 催化劑的制備
以六水合硝酸鎳[Ni(NO3)2·6H2O]為鎳源,采用等體積浸漬法制備催化劑。浸漬后樣品經(jīng)393 K干燥3 h后于馬弗爐中673 K焙燒3 h,并于流量為45 ml·min-1的氫氣中,在H2-TPR峰頂溫度 (Ni/ZrO2與Ni/SiO2催化劑在673 K還原,Ni/Al2O3催化劑在823 K還原) 恒溫還原3 h,得到Ni質(zhì)量分數(shù)為15%的催化劑,以ZrO2、SiO2及Al2O3為載體的催化劑分別標記為Ni/ZrO2、Ni/SiO2及Ni/Al2O3。
1.3 催化劑的評價
催化劑活性評價在100 ml不銹鋼高壓釜中進行。將0.1 g催化劑(0.25~0.42 mm)、4.9 g 順酐和40 ml 四氫呋喃加入反應釜中,密閉通氮氣置換出釜中的空氣后,充氫氣至一定壓力。待高壓釜加熱至反應溫度時,釜內(nèi)氫氣壓力增至反應所需壓力。此時,設定反應器攪拌轉(zhuǎn)速為400 r·min-1,開始反應。反應產(chǎn)物用HX-930型氣相色譜儀進行分析,毛細管色譜柱(φ 0.32 mm×25 m),固定液為5%甲基苯硅酮,柱溫353~473 K,程序升溫,氫離子火焰檢測器(FID)檢測各組分含量。
催化劑的套用實驗也是在上述實驗裝置上進行,當反應進行1 h時停止反應,將反應液過濾取出后,催化劑重新置于反應釜中,再次加入4.9 g順酐和40 ml的四氫呋喃,如此連續(xù)進行8次套用反應。
1.4 催化劑的表征
樣品的比表面積、孔體積和平均孔徑在 Micromeritics ASAP2020物理吸附儀上測定,采用靜態(tài)吸附法測定樣品的吸脫附等溫線,用BET (Brunauer-Emmett-Teller)方程計算樣品的比表面積。在Micromeritics 2920上進行樣品的H2-TPR表征,以5% H2/N2混合氣為載氣,用熱導池檢測器(TCD)檢測耗氫量。X射線衍射(XRD)采用 Bruker D8 Advance 型X射線衍射儀測定。Vantec檢測器,Cu靶,Kα(0.154 nm)輻射,管電壓40 kV,管電流40 mA。樣品的TPO-MS表征在NETZSCH STA449C 型熱分析儀上進行。以氧含量為15%(體積分數(shù))的O2-N2混合氣為載氣,流量為30 ml·min-1,升溫速率為10 K·min-1,反應生成的氣態(tài)混合物通過質(zhì)譜(HIDEN model QIC-20)在線檢測。
2 實驗結(jié)果與討論
2.1 催化劑的順酐液相加氫性能
2.1.1 氫氣壓力對催化劑順酐加氫性能的影響 表1列出了反應溫度為453 K時,不同氫氣壓力下各催化劑的評價結(jié)果。由表可知,各催化劑的順酐轉(zhuǎn)化率均在99.95%以上,順酐幾乎完全轉(zhuǎn)化。所有催化劑催化順酐加氫主產(chǎn)物均為丁二酸酐,另有少量γ-丁內(nèi)酯和丙酸生成,其余副產(chǎn)物如1,4-丁二醇、四氫呋喃等深度加氫產(chǎn)品的總選擇性在0.09%以下。據(jù)文獻報道[9-10,13-14],在順酐加氫反應中,順酐先經(jīng)鍵加氫生成丁二酸酐,然后丁二酸酐中的經(jīng)加氫/氫解生成γ-丁內(nèi)酯、丙酸等。在本文所考察的反應條件下,隨氫氣壓力升高,Ni/ZrO2與Ni/SiO2催化劑的γ-丁內(nèi)酯選擇性略有增加,當氫氣壓力由2 MPa升高至5 MPa時,Ni/ZrO2與Ni/SiO2催化劑的γ-丁內(nèi)酯選擇性分別由1.93%和0.85%升高至9.77%和4.26%。這表明高的氫氣壓力有利于丁二酸酐中的加氫生成γ-丁內(nèi)酯。Ni/Al2O3催化劑的丁二酸酐在99.65%以上,且隨氫氣壓力升高,Ni/Al2O3催化劑的γ-丁內(nèi)酯選擇性幾乎沒有變化,氫氣壓力對Ni/Al2O3催化劑加氫活性的促進作用并不明顯。
2.1.2 反應溫度對催化劑順酐加氫性能的影響 表2列出了氫氣壓力為5 MPa,在不同反應溫度時各催化劑的順酐加氫產(chǎn)物分布。反應溫度由453 K升高至483 K時,Ni/ZrO2與Ni/SiO2催化劑的γ-丁內(nèi)酯選擇性顯著增加,分別由9.77%與4.26%增至53.99%與39.87%。進一步提高反應溫度至513 K時,Ni/SiO2催化劑的γ-丁內(nèi)酯選擇性繼續(xù)增加至54.59%,Ni/ZrO2催化劑的γ-丁內(nèi)酯選擇性降至42.19%,其丙酸選擇性由2.43%增至11.81%,說明在高反應溫度下γ-丁內(nèi)酯進一步加氫生成丙酸[13]。Ni/Al2O3催化劑的γ-丁內(nèi)酯選擇性隨反應溫度升高略有增加,由453 K時的0.30%升高至513 K時的6.65%,但其丁二酸酐選擇性仍保持在90%以上。這些結(jié)果表明,提高反應溫度可顯著提高Ni/ZrO2與Ni/SiO2催化劑的加氫活性,而反應溫度同樣對Ni/Al2O3催化劑的γ-丁內(nèi)酯選擇性的提高非常有限,表明其幾乎沒有加氫活性。
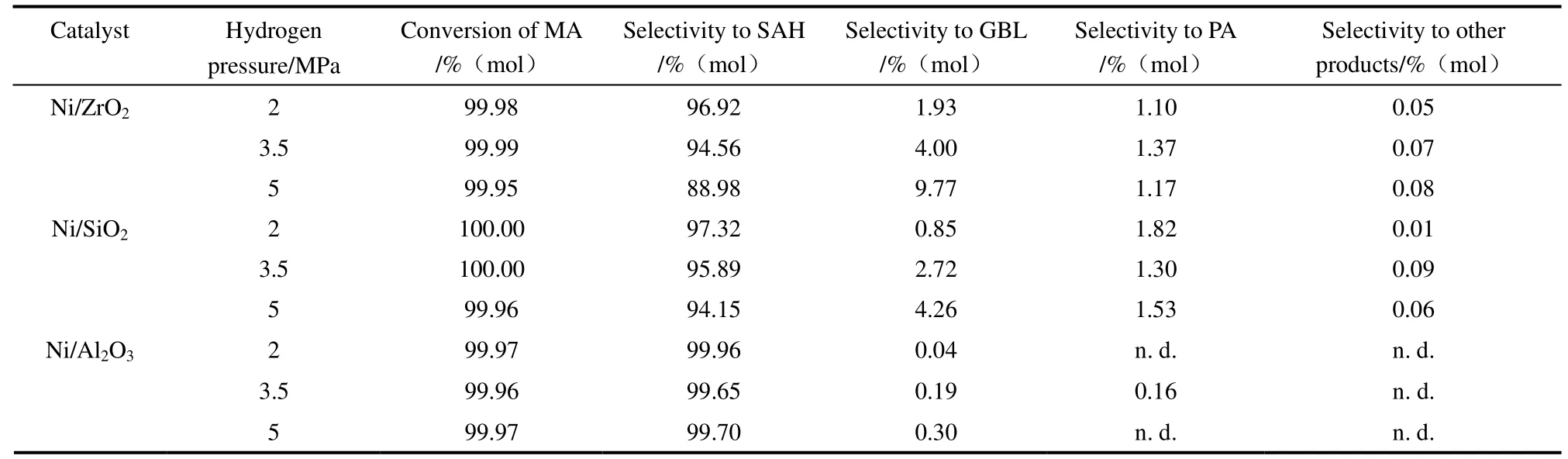
表1 氫氣壓力對催化劑順酐加氫性能的影響 Table 1 Effect of hydrogen pressure on catalytic performance of catalysts for maleic anhydride hydrogenation
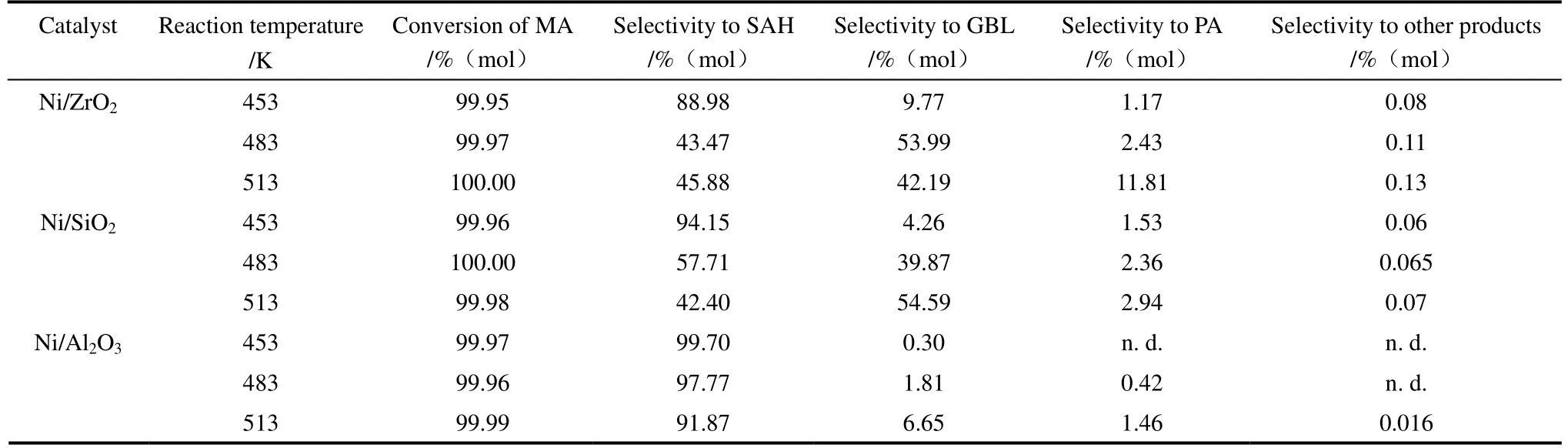
表2 反應溫度對催化劑順酐加氫性能的影響 Table 2 Effect of reaction temperature on catalytic performance of catalysts for maleic anhydride hydrogenation
2.1.3 反應時間對催化劑順酐加氫性能的影響 為進一步比較各催化劑的順酐加氫性能差異,圖1與圖2分別為反應溫度為453 K與483 K時反應時間對催化劑順酐加氫性能的影響。由圖1可知,各催化劑的順酐轉(zhuǎn)化率均隨反應時間的延長而逐漸增大。在反應時間相同時,Ni/Al2O3催化劑的順酐轉(zhuǎn)化率最高,Ni/SiO2催化劑最低。當反應時間為40 min時,Ni/Al2O3催化劑的順酐轉(zhuǎn)化率達到100%,而此時Ni/ZrO2與Ni/SiO2催化劑的順酐轉(zhuǎn)化率分別為83.20%與75.00%。這說明Ni/Al2O3催化劑具有最高的鍵加氫活性,能將順酐快速轉(zhuǎn)化為丁 二酸酐。由圖2中反應溫度為483 K時,反應時間由1 h延長至8 h時丁二酸酐與γ-丁內(nèi)酯選擇性的變化規(guī)律可知,隨反應時間延長,各催化劑的γ-丁內(nèi)酯選擇性逐漸增大,而丁二酸酐選擇性逐漸降低,證明在鎳基催化劑上γ-丁內(nèi)酯由丁二酸酐中的加氫制得,這與文獻報道結(jié)果一致[14]。值得關注的是,在所考察的催化劑中,Ni/ZrO2與Ni/SiO2催化劑的γ-丁內(nèi)酯選擇性均隨反應時間延長而顯著增加,至反應結(jié)束時,分別增至79.20%和60.00%。而Ni/Al2O3催化劑的γ-丁內(nèi)酯選擇性隨反應時間延長略有增加,至反應結(jié)束時,γ-丁內(nèi)酯選擇性僅為17.80%。
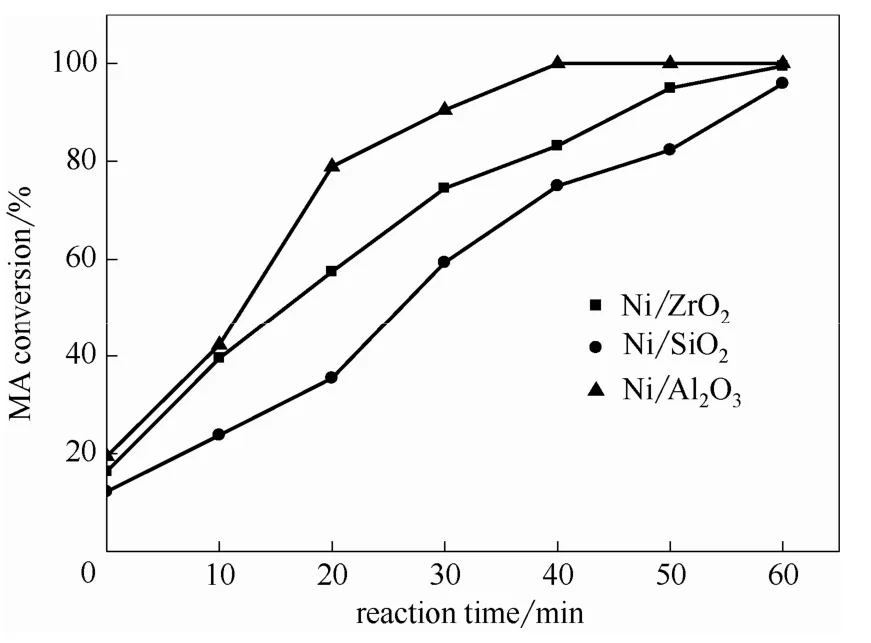
圖1 反應時間對催化劑順酐加氫性能的影響 Fig.1 Effect of reaction time on catalytic performance of catalysts for maleic anhydride hydrogenation (T= 453 K, PH2= 5 MPa)
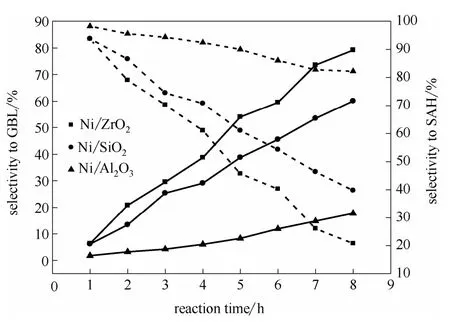
圖2 反應時間對催化劑順酐加氫性能的影響 Fig.2 Effect of reaction time on catalytic performance of catalysts for maleic anhydride hydrogenation (T= 483 K, PH2= 5 MPa)
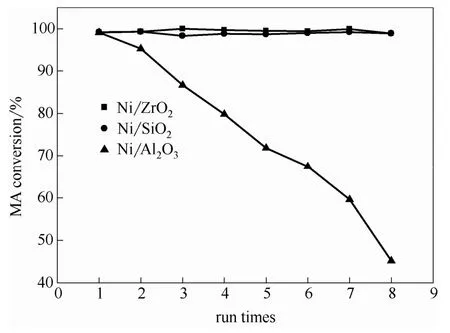
圖3 套用使用次數(shù)對催化劑順酐加氫性能的影響 Fig.3 Effect of run times on catalytic performance of catalysts for maleic anhydride hydrogenation (T= 483 K, PH2= 5 MPa, reaction time = 1 h)
2.1.4 催化劑的使用穩(wěn)定性 圖3為反應溫度為483 K、氫氣壓力5 MPa、反應時間為1 h時,隨套用使用次數(shù)增加,各催化劑順酐轉(zhuǎn)化率的變化趨勢。由圖可明顯地看到,在所考察的套用次數(shù)內(nèi),Ni/ZrO2與Ni/SiO2催化劑的順酐轉(zhuǎn)化率恒定在98.30%~100%,順酐轉(zhuǎn)化率沒有明顯的下降,具有良好的使用穩(wěn)定性。而Ni/Al2O3的順酐轉(zhuǎn)化率則急劇下降,使用穩(wěn)定性差。
由上述不同氫氣壓力、反應溫度、反應時間及套用次數(shù)下各催化劑的評價數(shù)據(jù)不難看出,Ni/Al2O3催化劑具有高的鍵加氫活性,可將順酐快速地加氫轉(zhuǎn)化為丁二酸酐,但其幾乎沒有加氫活性,其催化順酐加氫主產(chǎn)物為丁二酸酐。Ni/ZrO2催化劑的加氫活性最高,在反應溫度為453 K、氫氣壓力為5 MPa,反應時間8 h時,γ-丁內(nèi)酯選擇性達79.20%。此外,Ni/ZrO2與Ni/SiO2催化劑具有較好的使用穩(wěn)定性,而Ni/Al2O3催化劑則在使用過程中快速失活。
2.2 催化劑表征
2.2.1 BET表征 表3列出了催化劑比表面積、平均孔徑及孔容數(shù)據(jù)。由表中數(shù)據(jù)可知,不同載體制備催化劑的比表面積有較大差異,Ni/SiO2催化劑具有最高的比表面積,達507 m2·g-1,而Ni/ZrO2催化劑的比表面積最低,為83 m2·g-1。與催化劑比表面積相反,Ni/SiO2催化劑孔徑最小,為5.7 nm; 而Ni/ZrO2催化劑孔徑最大,為21.4 nm。除Ni/SiO2催化劑具有較大孔容外(0.73 cm3·g-1),Ni/ZrO2和Ni/Al2O3催化劑的孔容均較小。
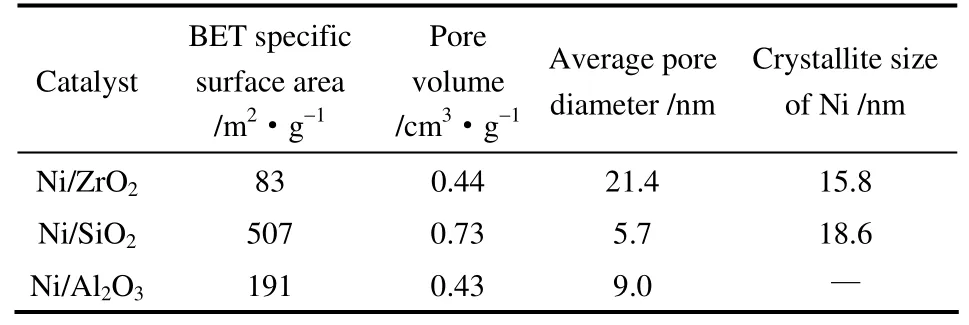
表3 不同載體負載鎳催化劑的織構參數(shù) Table 3 Texture parameter of supported nickel catalysts with different supports
2.2.2 XRD表征 還原后催化劑的XRD譜圖如圖4所示。Ni/SiO2與Ni/ZrO2催化劑均在2θ 為44.5°處呈現(xiàn)Ni(111)晶面[24-25]的特征衍射峰,其Ni晶粒的平均粒徑分別為18.6 nm和15.8 nm。與Ni/ZrO2及Ni/SiO2催化劑不同,Ni/Al2O3催化劑主要呈現(xiàn)出載體γ-Al2O3的特征衍射峰,僅在2θ為 45.8°處,呈現(xiàn)一個彌散的金屬Ni的衍射峰。這說明Ni/Al2O3催化劑中活性組分Ni幾乎以無定形存在。
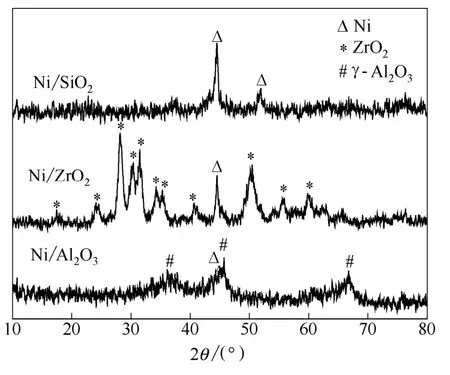
圖4 不同載體負載鎳催化劑的XRD譜圖 Fig.4 XRD patterns of supported nickel catalysts with different supports
2.2.3 H2-TPR表征 圖5為各催化劑的H2-TPR圖。由圖可知,Ni/SiO2催化劑在673 K出現(xiàn)一個主耗氫峰,同時在796 K左右有一個小的肩峰,表明在SiO2表面存在兩種不同狀態(tài)的NiO物種。位于673 K的低溫還原峰對應于與純NiO性質(zhì)類似的體相NiO的還原,高溫還原峰(796 K)則對應于與載體SiO2發(fā)生相互作用的NiO的還原[26]。Ni/ZrO2催化劑的H2-TPR峰是由630 K及663 K相互重疊的兩個主耗氫峰及763 K的次耗氫峰組成,這表明ZrO2表面NiO物種存在形態(tài)較為復雜。630 K和663 K處的耗氫峰分別對應于較小晶粒和較大晶粒體相NiO的還原;而763 K的高溫耗氫峰則歸屬為與載體具有較強相互作用的NiO物種的還原[27-28]。與Ni/ZrO2和Ni/SiO2催化劑的H2-TPR譜圖不同,Ni/Al2O3催化劑在約680 K至約1050 K表現(xiàn)出一個很寬化的耗氫峰,并在約723 K呈現(xiàn)一個很弱的肩峰。這表明大多數(shù)NiO與載體Al2O3具有很強的相互作用,NiO高度分散在Al2O3中。產(chǎn)生這一現(xiàn)象的原因可能是:在浸漬和焙燒過程中 Ni2+進入Al2O3的八面體及四面體空位生成了類鋁酸鎳物種,與Al2O3產(chǎn)生了強相互作用,即Al2O3將NiO鉚定在載體表面[29],有效阻止了Ni物種的聚集。
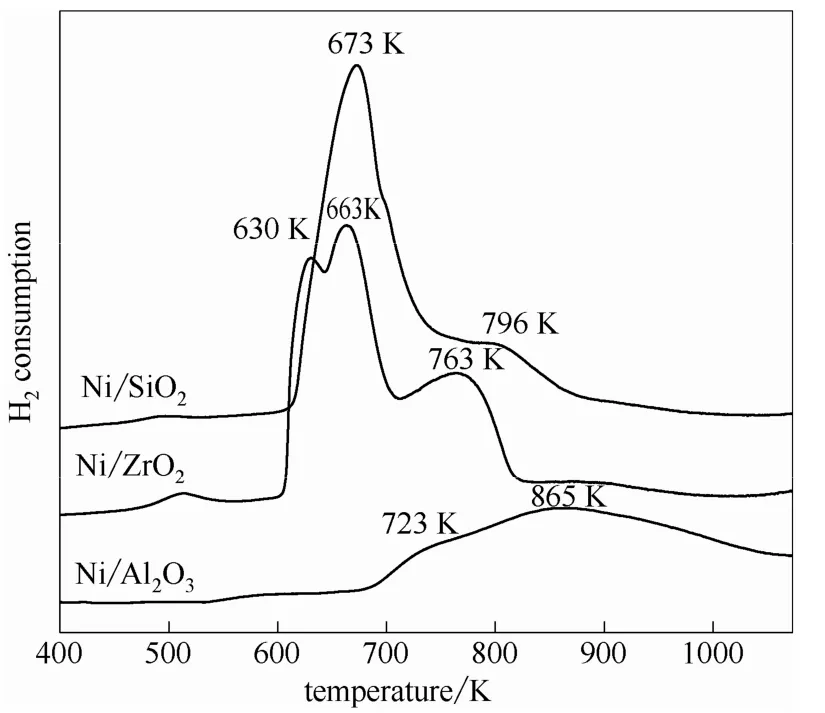
圖5 不同載體負載鎳催化劑的H2-TPR譜圖 Fig.5 H2-TPR profiles of supported nickel catalysts with different supports
綜合H2-TPR與XRD表征結(jié)果可以發(fā)現(xiàn),Ni/ZrO2與Ni/SiO2催化劑中,Ni前驅(qū)物主要以體相NiO存在。其中,Ni/ZrO2催化劑較Ni/SiO2催化劑具有較多的與載體具有相互作用的NiO物種,同時還有大量易還原的小粒徑NiO物種。而Ni/Al2O3催化劑中NiO與載體具有強的相互作用并高度分散于載體表面,還原后活性組分Ni以無定形存在。
一般認為,在金屬含量相同時,活性金屬的粒徑尺寸越小,活性金屬的比表面就越大,其提供的氫氣活化吸附位就越多,從而表現(xiàn)出高的加氫活性。本課題組的前期研究結(jié)果也表明,高分散、小晶粒尺寸的Ni基催化劑具有高的加氫活性即γ-丁內(nèi)酯選擇性[18-21]。Wang等[2]與Pillai等[3]關于Pd基催化劑催化順酐加氫的研究也得到了類似的結(jié)果:活性金屬Pd晶粒尺寸越小,其催化順酐加氫生成γ-丁內(nèi)酯的活性也愈高。Jeong 等[30]通過在Ni基催化劑中添加Pd、Mo助劑,減小活性金屬Ni晶粒尺寸,進而來提高催化劑合成γ-丁內(nèi)酯活性。Regenhardt等[14]關于不同載體負載鎳催化劑的順酐加氫性能研究表明,具有強金屬-載體相互作用的催化劑,活性金屬分散度高,表現(xiàn)出高的丁二酸酐加氫合成γ-丁內(nèi)酯性能。然而,與上述結(jié)論相悖的是,本工作中Ni/Al2O3催化劑活性金屬Ni物種以無定形態(tài)高度分散于Al2O3載體中,Ni/Al2O3催化劑的γ-丁內(nèi)酯選擇性卻遠低于Ni/ZrO2與Ni/SiO2催化劑,幾乎沒有加氫活性,且使用穩(wěn)定性很差。這可能是由于載體表面性質(zhì)不同,致使催化劑與反應物間存在不同的相互作用,進而使各催化劑的順酐加氫性能存在較大差異。
2.2.4 TPO-MS表征 為進一步認識影響各催化劑選擇性與使用穩(wěn)定性的原因,將套用實驗使用后的催化劑在15%O2-N2(體積分數(shù))混合氣中進行TPO-MS表征,同時,利用熱重在線監(jiān)測其質(zhì)量變化,得到的TG曲線如圖6所示。由圖可知,新鮮Ni/ZrO2催化劑在477~677 K的溫度范圍內(nèi)逐漸增重,這主要是由于活性金屬Ni在該溫度范圍內(nèi)被氧化為NiO所致。套用8次后Ni/ZrO2催化劑在563~628 K的溫度范圍內(nèi)呈現(xiàn)一個失重臺階(失重率約1.8%),而后又逐漸增重。其中較低溫度的失重臺階可歸屬為催化劑表面吸附有機物種的燃燒脫除[8],較高溫度的增重臺階則歸因于活性金屬Ni的氧化。套用使用后催化劑中活性金屬Ni的氧化溫度高于新鮮催化劑,可能是因為套用使用過程中催化劑表面吸附了有機物種,該有機物種覆蓋于活性金屬Ni表面,延遲了Ni的氧化。Ni/SiO2催化劑在309~394 K溫度區(qū)間呈現(xiàn)一明顯的失重臺階,歸屬為催化劑表面吸附水的脫除。另外,與Ni/ZrO2催化劑類似,Ni/SiO2催化劑在574~629 K溫度范圍內(nèi)緩慢失重(失重率約0.5%),而后又在629~704 K的溫度范圍內(nèi)逐漸增重,分別歸因于催化劑表面吸附有機物種的脫除和活性金屬Ni的氧化。與上述兩催化劑不同,Ni/Al2O3催化劑分別在300~440 K和440~750 K的溫度區(qū)間內(nèi)呈現(xiàn)兩個明顯的失重臺階,其中300~440 K間的失重率約4.1%,由催化劑表面吸附水的脫除引起,而440~750 K間由有機物燃燒脫除引起的失重率高達25.0%。這一結(jié)果說明,Ni/Al2O3催化劑在反應過程中吸附了大量有機 物種在其表面,這可能是該催化劑加氫活性以及使用穩(wěn)定性差的主要原因。
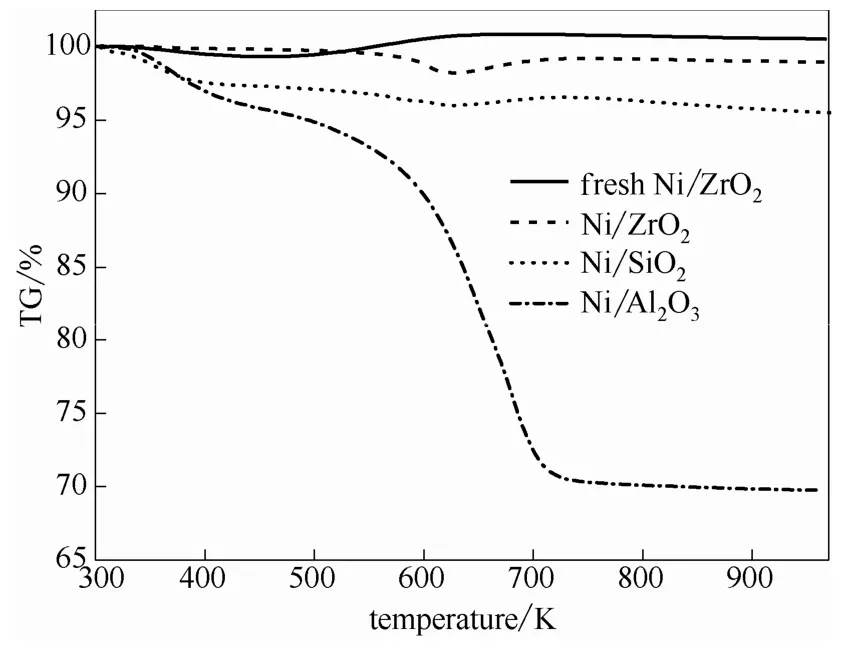
圖6 套用8次后催化劑的TG曲線 Fig.6 TG profiles of catalysts recycled for 8 times
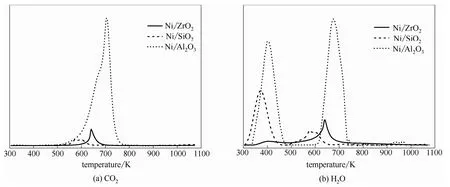
圖7 套用8次后催化劑的TPO-MS曲線 Fig.7 TPO-MS profiles of catalysts recycled for 8 times
圖7分別列出了使用后催化劑經(jīng)程序升溫氧化后監(jiān)測到的CO2和H2O曲線。由圖可知,Ni/SiO2、Ni/ZrO2與Ni/Al2O3催化劑分別在585、641、706 K處呈現(xiàn)一CO2峰,表明催化劑表面有機物種燃燒的難易程度不同,也進一步說明在催化劑套用使用過程中吸附于催化劑表面的有機物種不同。在與生成CO2相對應的溫度區(qū)間內(nèi),TPO-MS-H2O曲線上也呈現(xiàn)出相應的水峰。此外,Ni/SiO2與Ni/Al2O3的TPO-MS-H2O曲線上,還在較低溫度區(qū)間(300~440 K)呈現(xiàn)一水峰。這進一步證實,在TG曲線上,小于450 K的熱失重是由催化劑表面吸附水的脫除引起,而較高溫度的熱失重則是由催化劑表面吸附有機物種的燃燒脫除所致。
Yu等[6]認為,在金屬催化劑表面,吸附態(tài)順酐經(jīng)鍵加氫生成吸附態(tài)丁二酸酐,然后,丁二酸酐從催化劑表面脫附至反應體系中,反應體系中的丁二酸酐再次吸附到催化劑表面經(jīng)加氫后生成吸附態(tài)γ-丁內(nèi)酯,最后,γ-丁內(nèi)酯從催化劑表面脫附擴散至反應體系中。經(jīng)計算,Ni/SiO2、Ni/ZrO2與Ni/Al2O3催化劑表面有機物燃燒生成的CO2與H2O的物質(zhì)的量比分別約為2.7:1、2.6:1與6.4:1,即各催化劑表面吸附有機物種的碳氫比依次為1.35:1、1.3:1與3.2:1。這表明,Ni/SiO2與Ni/ZrO2催化劑在套用使用過程中,其表面吸附的有機物種較為一致,為順酐(碳氫比為2:1)與順酐加氫合成γ-丁內(nèi)酯的中間產(chǎn)物——丁二酸酐(碳氫比為1:1)的混合物。其中,Ni/ZrO2催化劑表面吸附有機物種含量為1.8%,而Ni/SiO2催化劑表面則僅有0.5%,說 明Ni/ZrO2催化劑具有較多的丁二酸酐吸附位點,這應該是Ni/ZrO2催化劑具有最高加氫活性的主要原因。與上述兩催化劑不同,Ni/Al2O3催化劑表面吸附的有機物種的量最高,達25.0%,約是Ni/SiO2催化劑的50倍,Ni/ZrO2催化劑的14倍,且該有機物種的碳氫比遠遠高于反應體系中任一物種的碳氫比(順酐的碳氫比最高,為2:1)。結(jié)合H2-TPR表征,推測這主要是由于Ni/Al2O3催化劑中載體表面存在較多八面體及四面體空位,這些八面體及四面體空位即為Al2O3表面的中強及強L酸酸性位點[31],可通過接受中氧原子的孤對電子而與丁二酸酐產(chǎn)生強的相互作用。在這一強相互作用下,丁二酸酐發(fā)生脫氫縮聚反應生成高碳物種覆蓋于催化劑表面,阻止了加氫反應的進行,顯著降低了Ni/Al2O3催化劑的加氫活性,表現(xiàn)出極低的γ-丁內(nèi)酯選擇性。在套用實驗過程中,越來越多的經(jīng)丁二酸酐脫氫縮聚形成的高碳物種覆蓋在活性金屬Ni表面,阻止了活性金屬Ni與氫氣或順酐的接觸,使Ni/Al2O3催化劑的加氫活性迅速降低。
3 結(jié) 論
ZrO2、SiO2與Al2O3負載Ni催化劑的順酐液相加氫產(chǎn)物主要是丁二酸酐和γ-丁內(nèi)酯。隨載體的不同,產(chǎn)物的選擇性有很大差異。順酐加氫至γ-丁內(nèi)酯中間產(chǎn)物——丁二酸酐與催化劑間的相互作用是影響產(chǎn)物選擇性及催化劑套用使用穩(wěn)定性的主要因素。由于ZrO2表面的特殊性質(zhì),使Ni/ZrO2催化劑表面具有較多的丁二酸酐吸附位點,這有利于丁二酸酐中的與活性金屬Ni發(fā)生加氫反應生成γ-丁內(nèi)酯。而Ni/Al2O3催化劑則與丁二酸酐存在強的相互作用,在這種強的相互作用下,丁二酸酐在該催化劑表面發(fā)生縮聚脫氫反應生成高碳物種,該高碳物種進一步覆蓋在Ni/Al2O3催化劑表面,阻止了加氫反應的進行,降低了Ni/Al2O3催化劑的活性,表現(xiàn)出極低的γ-丁內(nèi)酯選擇性,其催化順酐加氫的主要產(chǎn)物是丁二酸酐。在套用實驗中,越來越多的高碳物種覆蓋于活性金屬Ni表面,降低了Ni/Al2O3催化劑的使用穩(wěn)定性。
[1] Liao X, Zhang Y, Hill M, Xia X, Zhao Y, Jiang Z.Highly efficient Ni/CeO2catalyst for the liquid phase hydrogenation of maleic anhydride [J].Applied Catalysis A:General,2014, 488: 256-264.
[2] Wang Q, Cheng H, Liu R, Hao J, Yu Y, Zhao F.Influence of metal particle size on the hydrogenation of maleic anhydride over Pd/C catalysts in scCO2[J].Catalysis Today, 2009, 148: 368-372.
[3] Pillai U R, Sahle-Demessie E, Young D.Maleic anhydride hydrogenation over Pd/Al2O3catalyst under supercritical CO2medium [J].Applied Catalysis B:Environmental,2003, 43: 131-138.
[4] Xu J, Sun K, Zhang L, Ren Y, Xu X.A highly efficient and selective catalyst for liquid phase hydrogenation of maleic anhydride to butyric acid [J].Catalysis Communications, 2005, 6: 462-465.
[5] Jung S M, Godard E, Jung S Y, Park K C, Choi J U.Liquid-phase hydrogenation of maleic anhydride over Pd-Sn/SiO2[J].Catalysis Today,2003, 87: 171-177.
[6] Yu Y, Guo Y, Zhan W, Guo Y, Wang Y, Lu G.Effect of promoters on Cu-ZnO-SiO2catalyst for gas-phase hydrogenation of maleic anhydride to γ-butyrolactone at atmospheric pressure [J].Journal of Molecular Catalysis A:Chemical, 2014, 392: 1-7.
[7] Zhang D, Yin H, Xue J, Ge C, Jiang T, Yu L, Shen Y.Selective hydrogenation of maleic anhydride to tetrahydrofuran over Cu-Zn-M (M=Al, Ti, Zr) catalysts using ethanol as a solvent [J].Ind.Eng.Chem.Res., 2009, 48: 11220-11224.
[8] Meyer C I, Marchi A J, Monzon A, Garetto T F.Deactivation and regeneration of Cu/SiO2catalyst in the hydrogenation of maleic anhydride.Kinetic modeling [J].Applied Catalysis A:General, 2009, 367: 122-129.
[9] Gao C, Zhao Y, Liu D.Liquid phase hydrogenation of maleic anhydride over nickel catalyst supported on ZrO2-SiO2composite aerogels [J].Catalysis Letters,2007, 118: 50-54.
[10] Gao C, Zhao Y, Zhang Y, Liu D.Synthesis characterization and catalytic evaluation of Ni/ZrO2/SiO2aerogels catalysts [J].J.Sol-Gel Sci.Technol.,2007, 44: 145-151.
[11] Li J, Tian W, Wang X, Shi L.Nickel and nickel-platinum as active and selective catalyst for the maleic anhydride hydrogenation to succinic anhydride [J].Chemical Engineering Journal,2011, 175: 417-422.
[12] Feng Y, Yin H, Wang A, Xie T, Jiang T.Selective hydrogenation of maleic anhydride to succinic anhydride catalyzed by metallic nickel catalysts [J].Applied Catalysis A:General,2012, 425/426: 205-212.
[13] Meyer C I, Regenhardt S A, Marchi A J, Garetto T F.Gas phase hydrogenation of maleic anhydride at low pressure over silica-supported cobalt and nickel catalysts [J].Applied Catalysis A:General, 2012, 417/418: 59-65.
[14] Regenhardt S A, Meyer C I, Garetto T F, Marchi A J.Selective gas phase hydrogenation of maleic anhydride over Ni-supported catalysts: effect of support on the catalytic performance [J].Applied Catalysis A:General, 2012, 449: 81-87.
[15] Guo S, Shi L.Synthesis of succinic anhydride from maleic anhydride on Ni/diatomite catalysts [J].Catalysis Today,2013, 212: 137-141.
[16] Li J, Tian W, Shi L.Hydrogenation of maleic anhydride to succinic anhydride over Ni/HY-Al2O3[J].Ind.Eng.Chem.Res.,2010, 49: 11837-11840.
[17] Meyer C I, Regenhardt S A, Bertone M E, Marchi A J, Garetto T F [J].Gas-phase maleic anhydride hydrogenation over Ni/SiO2-Al2O3catalysts: effect of metal loading [J].Catal.Lett.,2013, 143: 1067-1073.
[18] Zhao Yongxiang(趙永祥), Qin Xiaoqin(秦曉琴), Hou Xicai(侯希才), Xu Xianlun(徐賢倫), Liu Diansheng(劉滇生).Preparation, characterization and properties of selective hydrogenation on Ni-based catalysts [J].Acta Phys.-Chim.Sin.(物理化學學報), 2003, 19: 450-454.
[19] Zhao Yongxiang(趙永祥), Wu Zhigang(武志剛), Xu Linping(許臨萍), Zhang Linqing(張臨卿), Liu Diansheng(劉滇生), Xu Xianlun(徐賢倫).Effect of nickel precursors on the catalytic performance of NiO/ SiO2[J].Acta Chimica Sinica(化學學報), 2002, 60: 596-599.
[20] Zhao Yongxiang(趙永祥), Wu Zhigang(武志剛), Xu Linping(許臨萍), Wang Yongzhao(王永釗), Liu Diansheng(劉滇生), Xu Xianlun(徐賢倫).A study on the property of NiO/SiO2aerogel catalysts (Ⅰ): Effect of nickel content and calcination temperature [J].Journal of Fuel Chemistry and Technology(燃料化學學報), 2001, 29: 178-181.
[21] Zhao Yongxiang(趙永祥), Wu Zhigang(武志剛), Wang Yongzhao(王永釗), Xu Linping(許臨萍), Liu Diansheng(劉滇生), Xu Xianlun(徐賢倫).Study on the property of NiO/SiO2aerogel catalysts (Ⅱ): Technological conditions of synthesis of γ-butyrolactone and succinic anhydride by maleic anhydride [J].Journal of Fuel Chemistry and Technology(燃料化學學報), 2001, 29: 182-184.
[22] Regenhardt S A, Trasarti A F, Meyer C I, Garetto T F, Marchi A J.Selective gas-phase conversion of maleic anhydride to propionic acid on Pt-based catalysts [J].Catalysis Communications, 2013, 35: 59-63.
[23] Vannice M A, Sen B.Metal-support effects on the intramolecular selectivity of crotonaldehyde hydrogenation over platinum [J].Journal of Catalysis, 1989, 115: 65-78.
[24] Gluhoi A C, M?rginean P, St?nescu U.Effect of supports on the activity of nickel catalysts in acetonitrile hydrogenation [J].Applied Catalysis A:General,2005, 294: 208-214.
[25] Bradford M C J, Vannice M A.Catalytic reforming of methane with carbon dioxide over nickel catalysts(Ⅰ): Catalyst characterization and activity [J].Applied Catalysis A:General, 1996, 142: 73-96.
[26] Xiong J, Chen J X, Zhang J Y.Liquid-phase hydrogenation of o-chloronitrobenzene over supported nickel catalysts [J].Catalysis Communications, 2007, 8: 345-350.
[27] Jasik A, Wojcieszak R, Monteverdi S, Ziolek M, Bettahar M M.Study of nickel catalysts supported on Al2O3, SiO2or Nb2O5oxides [J].Journal of Molecular Catalysis A:Chemical, 2005, 242: 81-90.
[28] Wu W, Xu J, Ohnishi R.Complete hydrodechlorination of chlorobenzene and its derivatives over supported nickel catalysts under liquid phase conditions [J].Applied Catalysis B:Environmental, 2005, 60: 129-137.
[29] Ren Shibiao(任世彪), Qiu Jinheng(邱金恒), Wang Chunyan(王春燕), Xu Bolian(許波連), Fan Yining(范以寧), Chen Yi(陳懿).Influence of nickel salt precursors on the hydrogenation activity of Ni/γ-Al2O3catalyst [J].Chinese Journal of Catalysis(催化學報), 2007, 28: 651-656.
[30] Jeong H, Kim T H, Kim K I, Cho S H.The hydrogenation of maleic anhydride to γ-butyrolactone using mixed metal oxide catalysts in a batch-type reactor [J].Fuel Processing Technology, 2006, 87: 497-503.
[31] Liu X.DRIFTS study of surface of γ-alumina and its dehydroxylation [J].Journal of Physical Chemistry C, 2008, 112: 5066-5073.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
