
不同結構茂金屬催化劑催化1-癸烯齊聚
2016-04-11 09:07:04高針針顧瑾璟米普科張藝鐔
石油煉制與化工 2016年11期
關鍵詞:催化劑
高針針,顧瑾璟,米普科,張藝鐔
(華東理工大學材料科學與工程學院,上海市先進聚合物材料重點實驗室,
不同結構茂金屬催化劑催化1-癸烯齊聚
高針針,顧瑾璟,米普科,張藝鐔
(華東理工大學材料科學與工程學院,上海市先進聚合物材料重點實驗室,
超細材料制備與應用教育部重點實驗室,上海 200237)
采用不同結構茂金屬催化劑催化1-癸烯齊聚反應,考察齊聚反應條件對1-癸烯齊聚及產物分布的影響。結果表明:不同結構的茂金屬對1-癸烯的催化活性和齊聚物組分分布影響顯著,非橋聯茂金屬、大位阻的茂金屬、限制構型的茂金屬以及雙核硅橋聯的茂金屬主要合成低黏度齊聚物(100 ℃運動黏度為2~5 mm2s,二聚體含量大于50%);Cs-對稱型茂金屬具有較高的催化活性,合成中等黏度的齊聚物(100 ℃運動黏度大于30 mm2s)。GC-MS分析結果表明,茂金屬催化合成的齊聚物主要由二聚體到五聚體的混合物組成。
茂金屬 1-癸烯 齊聚 結構性能關系
聚α-烯烴(PAO)是目前性能最佳的合成潤滑油基礎油,具有低黏度、高黏度指數、低揮發性、低流動性、較好的剪切性能及優良的抗高溫氧化性。合成PAO主要有均相及負載Lewis酸型、齊格勒-納塔型、茂金屬催化劑、鉻系催化劑、離子液體催化劑等[1]。但這些方法存在的問題是α-烯烴異構化嚴重,齊聚物相對分子質量分布寬,含有多個組分的低聚物以及高比例不適合作潤滑劑使用(容易揮發)的烯烴二聚體,基礎油黏度和黏度指數均不可控。茂金屬催化劑具有催化活性高、生成聚合物的相對分子質量分布窄、結構可控、相對分子質量可剪裁等優點[2]。利用茂金屬催化劑催化α-烯烴齊聚制備窄組成分布的PAO已進行了一些探索工作。(η5-RC5H4)2ZrCl2(R 為H,Me,i-Pr,n-Bu)[3-4]催化1-癸烯齊聚主要合成二聚體含量大于50%的齊聚物,分離出的1-癸烯二聚體與1-癸烯用陽離子催化劑催化合成特定的潤滑油基礎油。Kissin等[5]用(n-BuCp)2ZrCl2MAO(甲基鋁氧烷)分別催化1-己烯、4-甲基-1-戊烯、1-癸烯聚合,且其分離出的二聚體可用三氯化鋁固載催化劑和硅膠負載EtAlCl2催化劑進一步聚合,得到富含四聚體的PAO。Park等[6]制備了一種柄式茂金屬催化劑[(-C(Ph)H-C(Ph)H-)(η5-2,5-Me2C5H2)2ZrCl2]催化1-癸烯活性較高,合成的齊聚物適宜用作潤滑油基礎油。茂金屬催化α-烯烴合成PAO是一個具有潛力的領域[7-8],但是關于茂金屬催化劑結構與其催化α-烯烴聚合活性、齊聚物組成的構效關系尚不明確。本研究采用不同結構茂金屬催化劑催化1-癸烯齊聚反應,考察齊聚反應條件對1-癸烯齊聚及產物分布的影響。
1 實 驗
1.1 實驗原料
甲苯,分析純,上海凌峰化學試劑有限公司生產;二苯甲酮指示劑,金屬鈉回流24 h以上使用;1-癸烯,聚合級(95.8%),天津海納國際貿易有限公司生產,使用前柱層析純化,氫化鈣除水24 h;MAO,10%甲苯溶液,中國石油蘭州化工研究中心生產。
1.2 茂金屬催化劑
茂金屬催化劑,自制,分別制備非橋聯茂金屬(A)[9],大位阻非橋聯型茂金屬(B)[10],限制構型茂金屬催化劑(C)[11]、Cs-對稱型柄式茂金屬催化劑(D,E)[12],硅橋聯雙核茂金屬(F)[13]。
1.3 茂金屬催化1-癸烯齊聚
用氬氣將250 mL的三口燒瓶置換3~4次以除去氧氣和濕氣,將處理后且密封的50 mL 1-癸烯用注射器加入到反應器中,加熱到80 ℃,按一定AlZr摩爾比加入適量的MAO,然后加入含5 μmol茂金屬催化劑的甲苯溶液。反應6 h后用鹽酸乙醇溶液終止反應,蒸餾除去未反應的單體,計算單體轉化率。
1.4 齊聚物分析方法
2 茂金屬催化劑催化1-癸烯齊聚反應條件的考察
2.1 催化劑用量

圖1 C用量對1-癸烯齊聚反應的影響▲—單體轉化率; ■—運動黏度。 圖2、圖3同
在反應溫度為65 ℃、n(Al)∶n(Zr)=300的條件下,加入50 mL 1-癸烯反應6 h后,茂金屬催化劑C用量對1-癸烯齊聚反應的影響見圖1。由圖1可見,當C用量由3 μmol增加到5 μmol時,1-癸烯的單體轉化率逐漸增大,繼續增加C的用量,單體轉化率趨于穩定。這是由于隨著C用量逐漸增加,能引發聚合活性中心的數量也逐漸增加,所以1-癸烯的單體轉化率也隨之增大;隨著C用量的增加,催化活性位點增多,一定量的單體以較多的活性位點開始引發聚合,使得分子鏈變短,相對分子質量減小。從圖1還可以看出,隨著C用量由3 μmol增加到9 μmol,齊聚物100 ℃運動黏度由5.51 mm2s減小為2.79 mm2s。因此,選取C用量為5 μmol。
2.2 反應溫度
在C用量為5 μmol、n(Al)∶n(Zr)=300的條件下加入50 mL 1-癸烯反應6 h后,反應溫度對1-癸烯齊聚反應的影響見圖2。從圖2可以看出,當反應溫度由60 ℃升高至80 ℃時,1-癸烯單體轉化率逐漸增大,繼續升高溫度,單體轉化率有下降的趨勢。這是由于溫度過高,部分活性中心失活。隨著反應溫度的升高,利于β-H轉移終止和鏈轉移到助催化劑終止。從圖2還可以看出,反應溫度為60 ℃時,齊聚物100 ℃運動黏度為5.29 mm2s,當反應溫度升高到80 ℃時,齊聚物100 ℃運動黏度降為4.77 mm2s。因此,采用80 ℃作為聚合反應溫度。
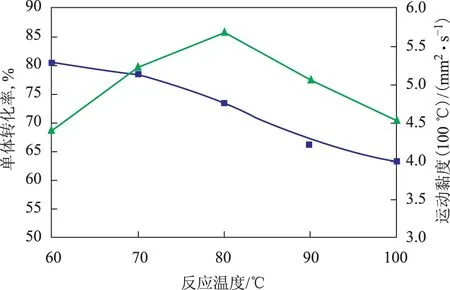
圖2 反應溫度對1-癸烯齊聚反應的影響
以MAO為助催化劑,在反應溫度為80 ℃、 C用量為5 μmol的條件下加入50 mL 1-癸烯反應6 h,考察AlZr摩爾比對1-癸烯齊聚反應的影響,結果見圖3。由圖3可見,當AlZr摩爾比為100時,單體轉化率僅為38.6%,AlZr摩爾比為300時,單體轉化率達到82.2%。這是由于AlZr摩爾比增加,MAO中的烷基鋁含量增加,使得1-癸烯的單體轉化率逐漸增大。從圖3還可以看出,隨著AlZr摩爾比從100增加到300,齊聚物100 ℃運動黏度由6.32 mm2s下降為3.97 mm2s。這是由于AlZr摩爾比增加利于鏈轉移到助催化劑終止,因此齊聚物的黏度會隨著AlZr摩爾比增加而下降。因此,選取AlZr摩爾比為300。

圖3 AlZr摩爾比對1-癸烯齊聚反應的影響
3 不同結構茂金屬催化劑對1-癸烯齊聚反應的影響
在反應溫度為80 ℃、茂金屬催化劑用量5 μmol、n(Al)∶n(Zr)=300的條件下加入50 mL 1-癸烯反應6 h,不同結構茂金屬催化劑對1-癸烯齊聚反應的影響見表1。從表1可以看出在A,B,C,D,E,F催化下的單體轉化率分別為82.5%,18.2%,84.0%,71.4%,77.5%,10.6%。A分子中的兩個茂環可以自由旋轉,活性中心空間位阻相對較小,利于1-癸烯的配位和插入;B分子中的茂環上含有兩個大體積取代結構配體,使得茂環只能做相對有限的旋轉,大體積取代基的空間位阻效應較高,阻礙了烯烴單體在活性中心的配位,使得1-癸烯插入困難,聚合活性較低;C分子中的碳橋聯限制構型咬合角α(Cp-Zr-N)僅為89.9°[14],完全可以看作是一個開放式結構,極大地擴展了聚合活性中心周圍的空間,單體可以在較大的空間里自由地配位-插入,但是給電子效應不如雙配體結構,結合兩方面原因,其催化活性稍高于非橋聯茂金屬A;Cs對稱的柄式橋聯茂金屬催化劑D、E,從空間位阻效應看,由于單碳原子的橋聯作用使咬合角α(Cp-Zr-Flu)縮小,活性中心配位空間相較于A更加開放,如催化劑D的α(Cp(centroid)-Zr-Flu)為118.6°[15],有利于長鏈1-癸烯在活性中心的插入、配位,因而具有較高的聚合活性,茂金屬催化劑D、E與強供電子基甲基、環己基相連,使得金屬活性中心更加穩定,利于烯烴單體的插入,硅橋聯雙核茂金屬催化劑F,相當于兩個Cp2ZrCl2通過剛性Me2Si橋聯起來,一個茂環和剛性Me2Si連接的一個Cp2ZrCl2相當于一個大體積的位阻,使得整個茂金屬催化劑只有有限的自由度,空間位阻效應增大,烯烴單體只能從一邊插入,因而其單體轉化率下降為10.6%。

表1 不同結構茂金屬催化劑對1-癸烯齊聚的影響
齊聚物黏度的不同主要是由于聚合機理不同造成的齊聚物分布不同。催化劑C催化1-癸烯合成齊聚物的GC-MS圖譜見圖4。由圖4可見,齊聚物主要是由二聚體到五聚體組成的混合物[16],這些齊聚物的結構規整性與陽離子催化劑引發1-癸烯聚合合成的齊聚物形成鮮明的對比,峰形較窄。茂金屬催化劑A~F催化1-癸烯合成齊聚物的組成分布見表2。從表1和表2可以看出:催化劑A催化1-癸烯合成的齊聚物的100 ℃運動黏度為2.75 mm2/s,所得齊聚物中二聚體含量為68.56%。這是因為α(cent(Cp)-Zr-Cent(Cp))之間的夾角為133°[17],也就是說在該催化劑中供聚合單體插入的空間相對較小,且催化活性高,當形成二聚體后,容易通過β-H消去或鏈轉移到助催化劑等使分子鏈斷裂,因此齊聚物中二聚體含量較高,多聚體較少;茂金屬B的茂環上連有較大的二苯基異丙基,大體積配體的強供電子效應使得金屬活性中心更加穩定,有利于鏈插入增長形成高聚體,因此相比A,其聚合物中二聚體含量降低為48.45%;限制構型茂金屬催化劑C合成的齊聚物二聚體含量約為51.37%,100 ℃運動黏度為4.26 mm2/s,這可能是由于該催化劑為限制構型,其咬合角Cp-Zr-N僅為89.9°,完全可以看作是一個開放式結構,單體可以在較大的空間里自由地配位-插入,其聚合物中二聚體含量下降很多,三聚體、四聚體、五聚體含量顯著上升,但由于聚合溫度較高,Al、Zr比較高,容易鏈轉移,使二聚體含量仍為主要部分;柄式茂金屬催化劑D,E均為Cs對稱型的柄式橋聯茂金屬,碳橋聯使得α(Cp(centroid)-Zr-Flu)變小,因此單體插入空間相對較大,該類型催化劑有高度的Cs對稱結構,利于烯烴單體的交替插入,且該類茂金屬催化劑均與強供電子基相連,使得金屬活性中心更加穩定,利于烯烴單體的插入,合成高聚物,柄式茂金屬催化劑合成的齊聚物黏度指數較高,產物性能較好,為理想的PAOs基礎油和黏度指數調節器;硅橋聯雙核茂金屬F催化合成的聚合物中二聚體含量降低為58.63%,而三聚體、四聚體含量略有升高,Mi等[13]發現這種硅橋聯雙核茂金屬催化劑催化烯烴合成的聚合物相對分子質量比催化劑A合成的聚合物相對分子質量高,他們歸因于金屬中心周圍的電子效應不同,由此看來電子效應對齊聚物的分布是非常重要的。從以上分析可以看出,用催化劑A,B,C,F主要合成低黏度齊聚物,用D,E合成的齊聚物黏度適中,黏度指數較高,可用作合成中黏度潤滑油基礎油。
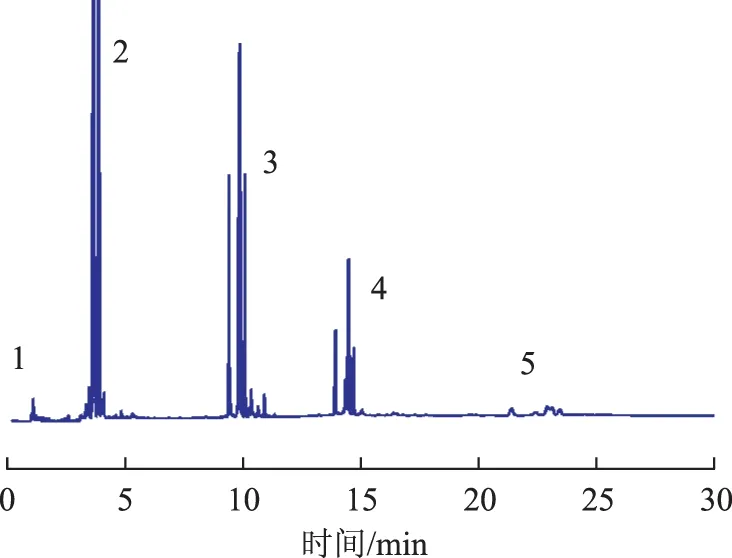
圖4 C催化1-癸烯合成齊聚物的GC-MS圖譜1—單體; 2—二聚體; 3—三聚體; 4—四聚體; 5—五聚體

催化劑齊聚物分布(w),%二聚體三聚體四聚體五聚體A68562223778143B484537601521124C513736491386172D1536339838941172E1277327643201127F58632785956397
4 1-癸烯齊聚物的結構與表征

圖5 C催化1-癸烯合成齊聚物的1H NMR圖譜
根據Carman和Wilkes提出的命名法辨別譜見圖6[21-22],參考和依據相關文獻給出13C NMR譜圖的碳標識和歸屬,化學位移在35.34,26.44,28.95,28.69,28.38,30.95,21.72,13.10處的側鏈峰分別歸屬于C1,C2,C3,C4,C5,C6,C7,C8,化學位移在120~140區間的峰歸屬于雙鍵上的碳。盡管實驗中的齊聚物并沒有加氫,所以含有一些不穩定的雙鍵,但是飽和及不飽和的齊聚物之間的黏度和黏度指數差距不大[23-24],因此仍有一定的研究意義。

圖6 C催化1-癸烯合成的齊聚物的13C NMR圖譜
5 結 論
(1) 限制構型茂金屬催化劑催化1-癸烯齊聚的最佳反應條件為:對于50 mL 1-癸烯,催化劑用量5 μmol;反應溫度80 ℃;AlZr摩爾比300,此時1-癸烯的單體轉化率為82.2%,齊聚物的100 ℃運動黏度為3.97 mm2s。
(2) 采用不同結構茂金屬催化劑催化1-癸烯齊聚,實驗發現茂金屬的結構對聚合活性及齊聚物分布影響較大。非橋聯茂金屬、大位阻的茂金屬、限制構型的茂金屬以及雙核硅橋聯的茂金屬主要合成低黏度齊聚物,Cs-對稱型茂金屬催化1-癸烯齊聚合成的齊聚物較為理想(適中的黏度,高的黏度指數和高的催化活性)。
(3) GC-MS 分析結果表明所得茂金屬合成的齊聚物混合物主要由二聚體、三聚體和四聚體、五聚體組成,且二聚體含量較多。
(4)1H NMR和13C NMR 圖譜顯示1-癸烯合成的齊聚物末端端基主要為乙烯基和三取代乙烯基,主要是通過β-H終止和β-H重排形成的,主要的鏈增長是通過1,2插入和2,1插入。
[1] 周在孝,丁洪生.α-烯烴齊聚制PAO催化劑的研究進展[J].應用化工,2011,40(11):2012-2014
[2] 王強,吳新君,李惠萍,等.烯烴聚合催化劑及其應用進展[J].石油化工,2004,33(S1):509-511
[3] Bagheri V,Eisenberg D C,Ratliff K S,et al.Oligomer oils and their manufacture:The United States,US6548723B2[P].2003-04-15
[4] Wu M M S,Coker C L.Process to produce low viscosity poly-alpha-olefins:the United States,US82017390[P].2012-06-26
[5] Kissin Y V,Schwab F C.Post-oligomerization of α-olefin oligomers:A route to single-component and multicomponent synthetic lubricating oils[J].J Appl Polym Sci,2009,111(1):273-280
[6] Park J H,Jang Y E,Jeon J Y,et al.Preparation of ansa-metallocenes for production of poly(α-olefin)lubricants[J].Dalton Trans.,2014,43(26):10132-10238
[7] K?hn R D,Haufe M,Kociok-K?hn G,et al.Selective trimerization of α-olefins with triazacyclohexane complexes of chromium as catalysts[J].Angew Chem Int Ed,2000,39(23):4337-4339
[8] Sattler A,Labinger J A,Bercaw J E.Highly selective olefin trimerization catalysis by a borane-activated titanium trimethyl complex[J].Organometallics,2013,32(23):6899-6902
[9] 向文軍,陳發德,姜振華,等.二氯二茂鋯的合成及其對丁苯共聚物加氫的催化活性[J].合成化學,2010,18(4):484-486
[10]許勝,黃吉玲.大取代茂鋯配合物合成及分子內消除反應研究[J].化學學報,2005,63(14):1318-1322
[11]Mi Puke,Kong Xiaojuan,Xu Sheng,et al.Synthesis of sp3C1-bridged constrained geometry complexes and their application for copolymerization of ethylene and 1-octene[J].Adv Mater Res,2012,683:280-288
[12]Ewen J A,Jones R L,Razavi A,et al.Syndiospecific propylene polymerizations with Group ⅣB metallocenes.[J].J Am Chem Soc,1988,110(18):6255-6256
[13]Mi Puke,Xu Sheng,Qu Liangduan.Synthesis of double silylene-bridged binuclear metallocenes and their cooperation effect during ethylene polymerization and ethylene1-hexene copolymerization[J].J Appl Polym Sci,2011,121:21-26
[14]Jesus C,Klaus K.How to synthesize a constrained geometry catalyst(CGC)—A survey[J].J Organomet Chem,2007,692:4411-4423
[15]Wang Baiquan.Ansa-metallocene polymerization catalysts:Effects of the bridges on the catalytic activities[J].Coordination Chemistry Reviews,2006,250(12):242-258
[16]盛亞平,黃啟谷,陳偉,等.茂金屬催化劑/MAO催化1-癸烯齊聚合及其產物的結構與性能[J].化工學報,2007,58(3):759-764
[17]Cerrada M L,Benavente R,Pérez E,et al.Ethylene10-undecenoic acid copolymers prepared with different metallocene catalysts[J].Macromol Chem Phys,2007,208(8):841-850
[18]Shao Huaiqi, Li Hui,Lin Jichao,et al.Metallocene-catalyzed oligomerizations of 1-butene and α-olefins:Toward synthetic lubricants[J].European Polymer Journal,2014,59:208-217
[20]Kim I,Zhou J M,Chung H.Higher α-olefin polymerizations catalyzed by rac-Me2Si(1-C5H2-2-CH3-4-tBu)2Zr(NMe2)2Al(iBu)3[Ph3C][B(C6F5)4][J].J Polym Sci Part A:Polym Chem,2000,38(9):1687-1697
[21]Vinceozno B,Roberta C.Microstructure of polypropylene[J].Prog Polym Sci,2001,26(3):443-533
[22]Tanaka H,Harada H.13C NMR spectra of isotactic polypropylen film during stretching and shrinkage[J].Eur Polym J,1991,27(1):1-5
[23]Grigor’eva N G,Bubennov S V,Kutepov B I.Oligomerization of 1-octene catalyzed by zeolites[J].Catal Ind,2011,3(2):144-150
[24]Li Deng,Shen Benxian,Sun Hui.Oligerization of 1-decene:Catalyzation by immobilized AlCl3γ-Al2O3catalyst in fixed-bed reactor[J].China Petroleum Processing & Petrochemical Technology,2014,16(3):50-55
OLIGOMERIZATION OF 1-DECENE WITH DIFFERENT STRUCTURE METALLOCENE CATALYSTS
Gao Zhenzhen, Gu Jinjing, Mi Puke, Zhang Yixin
(1.ShanghaiKeyLaboratoryofAdvancedPolymericMaterials,KeyLaboratoryforUltrafineMaterialsofMinistryofEducation,SchoolofMaterialsScienceandEngineering,EastChinaUniversityofScienceandTechnology,Shanghai200237)
The oligomerization of 1-decene was catalyzed by metallocene catalysts with different structures and the effect of conditions on 1-decene oligomerization reaction and properties of oligomers were invesgated as well. It is found that the structure of metallocene catalysts affects the catalytic behavior and the distribution of oligomer components significantly. The unbridged metallocene catalysts, sterically hindered metallocene catalysts, constrained geometry configuration metallocene catalysts, and silicon-bridged binuclear metallocene catalysts mainly produce low viscosity oligomers (kinematic viscosity at 100 ℃: 2—5 mm2s) with dimer content more than 50%); it is interested to find thatCssymmetrical metallocene catalysts show a higher activity and primarily produce medium-high viscosity oligomers (kinematic viscosity at 100 ℃>30 mm2s). The GC-MS results demonstrate that the oligomer mixture is composed of dimer to pentamer.
metallocene; 1-decene; oligomerization; structure-property relations
2016-03-17; 修改稿收到日期: 2016-07-25。
高針針,碩士,主要從事烯烴聚合研究工作。
米普科,E-mail:mpk869@163.com。
國家自然科學基金項目(U1162111,U1362111)。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
