NO在Pt摻雜Rh(111)表面吸附的理論研究
2016-06-05 15:00:58張福蘭解曉華胡武洪徐伯華
四川師范大學學報(自然科學版) 2016年5期
張福蘭,解曉華,胡武洪,徐伯華
(1.長江師范學院化學化工學院,重慶408003; 2.重慶市無機特種功能材料重點實驗室,重慶408003)
NO在Pt摻雜Rh(111)表面吸附的理論研究
張福蘭1,2,解曉華1,2,胡武洪1,2,徐伯華1,2
(1.長江師范學院化學化工學院,重慶408003; 2.重慶市無機特種功能材料重點實驗室,重慶408003)
采用密度泛函理論與周期性平板模型相結合的方法,對NO在Rh(111)表面top、fcc、hcp、bridge 4個吸附位和Rh-Pt(111)表面Rh-top、Pt-top、RhRh-bridge、RhPt-bridge、PtPt-bridge、Rh2Pt-hcp、RhPt2-hcp、Rh2Pt-fcc、RhPt2-fcc 9個吸附位的13種吸附模型進行了構型優化、能量計算,得到了NO較有利的吸附位;并對最佳吸附位進行總態密度分析.結果表明:NO在Rh(111)和Rh-Pt(111)表面的最穩定吸附位分別為Rh-top和Pt-top位,其吸附能的大小順序為Ph(111)<Rh-Pt(111);NO與金屬表面成鍵,屬于化學吸附.
NO;Rh(111)表面;金屬摻雜;態密度;密度泛函理論
機動車尾氣的排放是大氣污染的一個重要來源.城市交通系統產生的大氣污染物主要包括CO、NOx、非甲烷碳氫化合物THC以及其他有害物質以及可吸入顆粒污染物.據此,迫切需要對機動車尾氣進行凈化處理.但目前機動車尾氣凈化催化劑的抗老化和耐久性能較差[1-2].為了找到還原性好、儲氧量高、將儲氧、空燃比特性、空速特性及溫度特性最優的催化劑,研究者們做了大量工作[3-7].近幾年,對于機動車尾氣凈化的催化劑相關研究多是鑒于實驗研究[3-8],理論研究相對甚少[9].本研究小組曾采用密度泛函理論與周期性平板模型相結合的方法,多次研究過小分子在金屬表面的吸附性能[10-14],并得到了很好的研究結果.因此,為了解決目前機動車尾氣凈化催化劑方面存在的相關問題,也為了彌補相關研究的理論空缺,運用密度泛函理論和周期平板模型相結合的方法,首先模擬了相同Rh原子數目的(100)、(110)、(111)3個低指數面,發現Rh(100)表面穩定化能比Rh(110)表面穩定化能低3.34 eV,Rh(111)表面穩定化能比Rh (110)表面穩定化能低5.61 eV,即3個表面的穩定化能關系為 ERh(111)<ERh(100)<ERh(110),所以 Rh (111)表面最穩定.所以,選取了能量最低的Rh (111)為催化劑表面,模擬研究了NO在Rh(111)和Rh-Pt(111)表面的吸附行為,為找尋出活性最高的催化劑,為開發造價低、效率高、更環保實用的機動車尾氣凈化催化劑提供一定的理論依據.
1 計算模型與方法
采用密度泛函理論和周期性平板模型相結合的方法模擬計算了NO在Rh(111)和Rh-Pt(111)表面的吸附情況.采用類似A.Michaelides等[15]研究CH2和H在Ni(111)表面吸附模型,取3×3×5 5層原子的平板周期性模型模擬Rh(111)表面,而Rh-Pt(111)表面是在Rh(111)表面基礎之上按物質的量NRh∶NPt=1∶1的比例摻雜Pt模擬得到,如圖1所示,(a)、(b)分別是 Rh(111)和 Rh-Pt(111)的真空周期表面模型的俯視圖及其NO在Rh(111)、Rh-Pt(111)表面的吸附位,真空層厚度為2.0 nm.計算過程中考慮表面弛豫,固定下面3層原子,讓與吸附物種最近2層金屬原子弛豫;采用DNP基組;對N和O原子采用全電子基組,而對Rh和Pt原子采用有效核勢(ECP)贗勢;函數采用廣義梯度近似(GGA)中 Perdew-Wang-91 (PW91)函數;Quality設為fine,優化收斂精度取程序內定值.吸附能通過下面方程計算得到其中,Eads和Es分別代表吸附前NO和Rh(111)、Rh-Pt(111)的能量,Eads/s代表吸附NO后體系的總能量.全部計算工作使用MS 5.0版本中的Dmol3模塊[16-17].

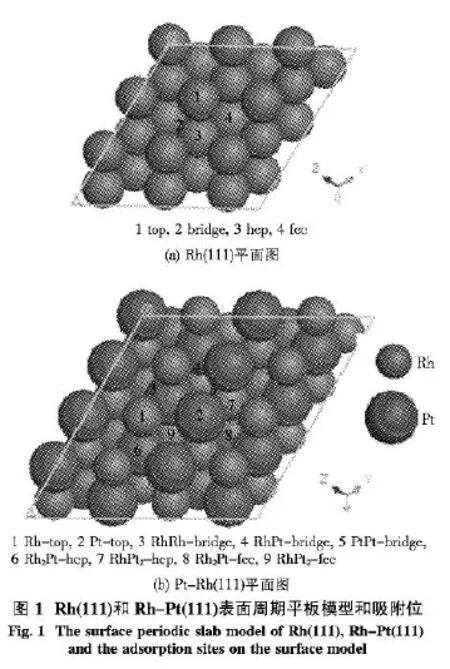
2 結果與討論
2.1 NO在Rh(111)和Rh-Pt(111)表面吸附的平衡構型 NO在Rh(111)和Rh-Pt(111)表面各吸附位的吸附能為:Rh(111)的top、bridge、hcp、fcc位分別為2.32、2.15、1.88、2.19 eV;Rh-Pt(111)的Rh-top、Pt-top、RhRh-bridge、RhPt-bridge、PtPt-bridge、Rh2Pt-hcp、RhPt2-hcp、Rh2Pt-fcc、RhPt2-fcc位分別為3.44、3.64、2.74、3.45、3.23、3.46、3.44、3.44、3.45 eV.圖2是NO以及NO吸附在Rh(111)和Rh-Pt(111)表面最佳吸附位的構型參數.在研究過程中,首先優化了 NO分子、Rh(111)和Rh-Pt(111)表面,計算得到氣相NO分子中N—O鍵長為0.116 3 nm,構型參數見圖2(a).然后,將優化好的NO分子垂直放在優化過的Rh(111)和Rh-Pt(111)表面的真空層中進行第2次優化,最后得到了NO分子吸附到Rh(111)和Rh-Pt(111)表面的穩定構型.
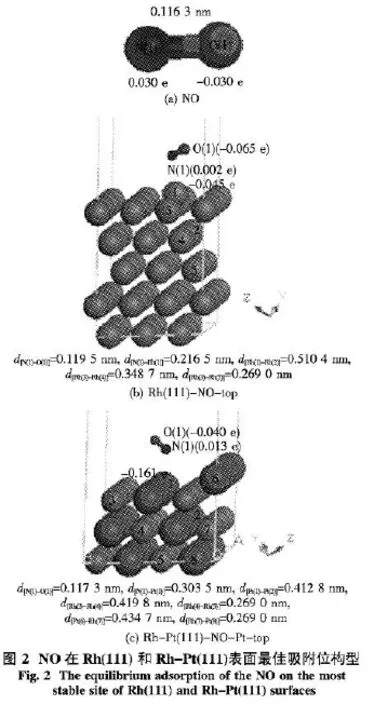
研究表明,優化過程中,NO在Rh(111)表面發生偏轉,穩定后不再是垂直狀態,而是與金屬表面保持大約30°的角度,而且NO在Rh(111)表面top、bridge、hcp、fcc 4個位中top位的吸附能絕對值最大,為-2.32 eV,所以top是最穩定吸附位,top位構型參數見圖2(b).當NO吸附在Rh(111)表面top位時,N—O鍵的鍵長被拉長了0.003 2 nm,說明N—O鍵被活化.密立根電荷分析表明,NO吸附在Rh(111)面時,金屬表面向NO分子轉移了0.063 e.優化時,考慮表面弛豫,固定了下面3層Rh原子,讓離NO最近的2層原子自由移動.由圖2(b)
中 數 據 可 知,d[Rh(3)-Rh(4)]= 0.348 7 nm,d[Rh(4)-Rh(5)]=0.269 0 nm,說明 Rh的上面2層原子分別弛豫了0.079 7 nm;又對比d[Rh(1)-Rh(2)]= 0.510 4 nm 和 d[Rh(3)-Rh(4)]=0.348 7 nm,說明Rh(1)原子弛豫更嚴重,這是由于Rh(1)原子與NO分子成鍵所導致的結果.
又通過計算比較NO在Rh-Pt(111)表面9個位的吸附能發現Pt-top位的吸附能-3.64 eV的絕對值最大,為最佳吸附位,構型參數見圖2(c).比較發現,摻雜Pt后,NO在催化劑表面的吸附能增強,這是由于Rh(111)的d軌道更有利于向NO的π*反鍵軌道反饋電子.與圖2(b)對比,NO吸附在Rh-Pt(111)表面Rt-top位達到穩定構型時,NO也發生了偏轉,與 Rh-Pt(111)表面成大約30°,N(1)到Pt(1)原子的距離增加為0.303 5 nm,同時N—O鍵由0.116 3 nm拉長到0.117 3 nm,說明N—O鍵略被削弱,由金屬表面轉移的電子數減少,這點從Rt(1)原子所帶的電荷為-0.161 e,比Rh(111)表面的Rh(1)原子所帶的電荷為-0.045 e更負也能得到說明.由圖 2(c)中數據可知,d[Rh(3)-Rh(4)]=0.419 8 nm,d[Rh(4)-Rh(5)]=0.269 0 nm,說明 Rh原子表面弛豫了 0.150 8 nm; d[Pt(6)-Rh(7)]=0.434 7 nm,d[Rh(7)-Pt(8)]=0.269 0 nm,說明Pt原子表面弛豫了0.165 7 nm.又對比圖2(b)中數據可知,當Rh表面摻雜Pt原子后,表面弛豫更嚴重.這個結論與NO吸附在Rh(111)和Rh-Pt(111)時,結合能分別為-2.32和-3.64 eV,依次增強的結論非常吻合.所以結論是可靠的.
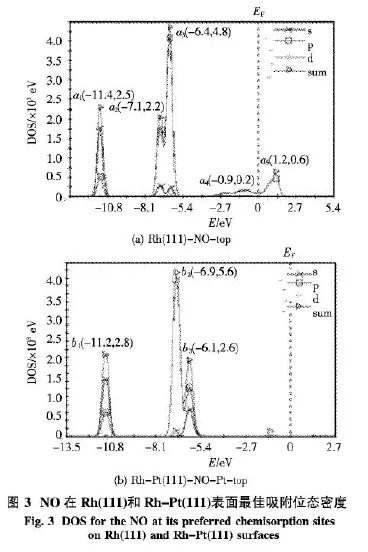
2.2 態密度(DOS)分析 圖3給出了 NO在Rh(111)和Rh-Pt(111)表面的最佳吸附位的態密度(DOS)圖,其中(a)為NO在Rh(111)面top位的態密度圖,(b)為NO在Rh-Pt(111)面Pt-top位的態密度圖.根據NO自由分子的分子軌道排布式分析,圖3(a)中在低于費米能級(EF)以下大約11.4 eV處的a1是NO的(σ2s)2軌道能級,它是由O原子的2s-2p相互混雜的分子軌道能級,但主要貢獻在2s;在低于EF以下大約7.1 eV處的a2是NO的(σ2p)2軌道能級,它是由N和O原子的2s-2p形成的分子軌道能級,但主要貢獻在2p;在低于EF以下大約6.4 eV處的a3是NO的成鍵二重簡并能級(π2p)4,是由2p-2p形成的分子軌道能級;從EF以下大約3.0 eV直至以上大約2.5 eV一連續寬峰是NO與金屬表面形成的σ—π配鍵,其中a4是NO的軌道和Rh的空雜化軌道形成的Rh←NO σ配鍵,a5是NO空的反鍵軌道和Rh的4d形成的Rh→NO反饋π鍵.yz
對比圖3(a)和(b)發現,(b)中的b1特征峰與(a)中的a1特征峰類似,只是能量和DOS略有差別,見圖3中各個峰頂括號中數據;而(b)中的b2、b3兩組特征峰分別類似于與(a)中的a2、a32組特征峰,其中在低于EF以下大約6.9 eV處的b2是NO的N和O原子的2s-2p和2p-2p形成的分子軌道能級,但主要貢獻在2p;在低于EF以下大約6.1 eV處的b3是NO的成鍵二重簡并能級(π2p)4,是由2p-2p形成的分子軌道能級;而摻雜Pt后,NO與金屬表面沒有形成的σ—π配鍵,從數據顯示,態密度強度降低了很多,這與吸附能降低結論一致,也與前面所得摻雜Pt后金屬表面原子所帶電荷更負結論一致.
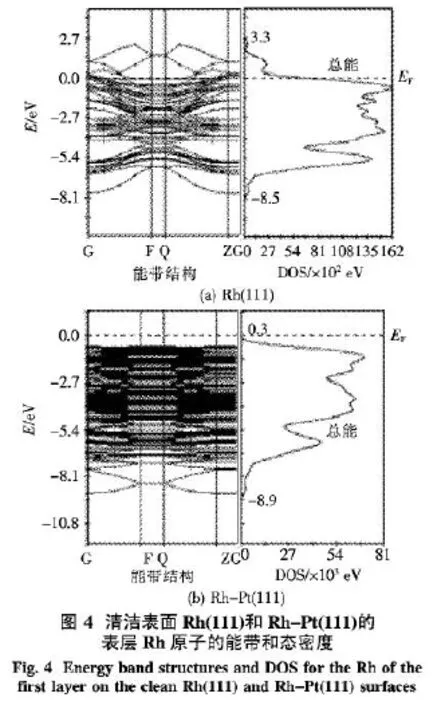
圖4給出了清潔表面 Rh(111)和 Rh-Pt (111)的表層Rh原子的能帶和態密度(DOS)圖.由圖4可知,Rh(111)表面的能帶區域為-8.5~3.3 eV,Rh-Pt(111)表面的能帶區域為-8.9~0.3 eV,Rh金屬摻雜Pt后,Rh催化劑的能帶區變窄了3.2 eV,而且向費米能級以下移動,所以,Rh摻雜Pt后,催化效果更好.
3 結論
采用密度泛函理論與周期性平板模型相結合的方法,對NO在Rh(111)表面4個吸附位和Rh-Pt(111)表面9個吸附位的13種吸附模型進行了構型優化、能量計算,得到了NO較有利的吸附位;并對最佳吸附位進行總態密度分析.結果表明:NO在Rh(111)和Rh-Pt(111)表面的最穩定吸附位分別為Rh-top和Pt-top位,其吸附能的大小順序為Rh(111)<Rh-Pt(111);NO與金屬表面成鍵,屬于化學吸附.
致謝 重慶市無機特種功能材料重點實驗室項目(KFKT201506)對本文給予了資助,謹致謝意.
[1]盛葉琴,周瑛,盧晗鋒,等.鈰基復合氧化物催化碳煙燃燒的性能及其H2-TPR研究[J].催化學報,2013,34(3):567-577.
[2]DATURI M,FINOCCHIO E,BINET C,et al.Reduction of high surface area CeO2-ZrO2mixed oxides[J].J Phys Chem,2000,B104:9186-9194.
[3]SHANG H Y,WANG Y,GONG M C,et al.Pd catalysts supported on modified Zr0.5Al0.5O1.75used for lean-burn natural gas vehicles exhaust purification[J].J Nat Gas Chem,2012,21(4):393-399.
[4]RAINER D R,KORANNE M,VESECKY S M,et al.CO+O2and CO+NO reactions over Pd/Al2O3catalysts[J].J Phys Chem,1997,B101(50):10769-10774.
[5]WANG Q Y,LI G F,ZHAO B,et al.Investigation on properties of a novel ceria-zirconia-praseodymia solid solution and its application in Pd-only three-way catalyst for gasoline engine emission control[J].Fuel,2011,90(10):3047-3055.
[6]崔亞娟,何勝楠,方瑞梅,等.整體式Pd/La2O3-Al2O3和Pd/CeO2-ZrO2-Y2O3催化劑上汽油車尾氣凈化性能的比較[J].催化學報,2012,33(6):1020-1026.
[7]王蘇寧,崔亞娟,蘭麗,等.一個新的滿足未來排放標準的摩托車尾氣凈化催化劑[J].催化學報,2014,35(9):1482-1491.
[8]李明利,余瓊,周春江,等.單鈀型汽車尾氣凈化催化劑的催化活性研究[J].兵器材料科學與工程,2012,35(5):35-37.
[9]蘇海燕,李微雪,包信和.Pt(111)和Pt3Ni(111)表面上CO催化氧化反應的密度泛函理論研究[J].催化學報,2008,29(8):683-686.
[10]張福蘭,李來才.CO在Pt(111)和Pt-M(111)(M=Ni,Mg)表面吸附的理論研究[J].分子科學學報,2011,27(1): 49-53.
[11]張福蘭.C2Hx(x=4~6)在Co(111)表面吸附的理論研究[J].四川師范大學學報(自然科學版),2012,35(5): 686-689.
[12]張福蘭.N2在Co摻雜Ru(001)表面吸附的DFT研究[J].原子與分子物理學報,2010,27(4):769-774.
[13]張福蘭.CHx(x=2~4)在Fe(110)表面吸附的DFT研究[J].原子與分子物理學報,2010,27(5):986-992.
[14]張福蘭.C2Hx(x=4~6)在Ni(111)表面吸附的DFT研究[J].原子與分子物理學報,2010,27(6):1175-1182.
[15]MICHAELIDES A,HU P.A density functional theory study of CH2and H adsorption on Ni(111)[J].J Chem Phys,2000,112(13):6006.
[16]DELLEY B.An all-electron numerical method for solving the local density functional for polyatomic molecules[J].J Chem Phys,1990,92(1):508.
[17]DELLEY B.From molecules to solids with the DMol3approach[J].J Chem Phys,2000,113(18):7756.
Theoretical Study of NO Adsorption on Rh(111)Surface by Doping with Pt
ZHANG Fulan1,2,JIE Xiaohua1,2,HU Wuhong1,2,XU Bohua1,2
(1.College of Chemistry and Chemical Engineering,Yangtze Normal University,Chongqing 408003; 2.Chongqing Key Laboratory of Inorganic Special Functional Materials,Yangtze Normal University,Chongqing 408003)
The density functional theory(DFT)and periodic slab model have been used to investigate NO adsorption on Rh(111) and Rh-Pt(111)surfaces.The adsorption energies,equilibrium geometries of NO on four possible sites(top,fcc,hcp,and bridge)on Rh(111)surface and on 9 possible sites[Rh-top,Pt-top,RhRh-bridge,RhPt-bridge,PtPt-bridge,Rh2Pt-hcp,RhPt2-hcp,Rh2Ptfcc,RhPt2-fcc]on Rh-Pt(111)surface have been predicted and compared.The relatively favorable adsorption sites have been found.Density of states analysis of the most stable sites have been discussed.The results reveal that the most stable sites for NO adsorption on Rh(111)and Rh-Pt(111)surfaces are Rh-top and Pt-top,respectively;the order of the adsorption energy on the most stable sites is Ph(111)<Rh-Pt(111).The calculated results show that there are chemical bonds between NO and Rh(111),Rh-Pt(111)surface,respectively.
nitron monoxide;Rh(111)surface;metal doped;density of states;density functional theory
O641;O647
A
1001-8395(2016)05-0730-05
10.3969/j.issn.1001-8395.2016.05.020
(編輯 周 俊)
2015-08-22
重慶市教委科學技術項目(KJ1601215,KJ1401227和KJ15012002)和重慶市涪陵區科技計劃項目(FLKJ2015ABA1042)
張福蘭(1976—),女,副教授,主要從事應用量子化學的研究,E-mail:cjsy0606@163.com
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
汽車工程學報(2017年2期)2017-07-05 08:13:02
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
