第八章環境催化
2016-07-25 01:47:53李俊華
工業催化 2016年5期
關鍵詞:催化劑
李俊華,賀 泓
(1.清華大學環境學院,北京 100084; 2.中國科學院生態環境研究中心,北京 100085)
?
第八章環境催化
李俊華1,賀泓2
(1.清華大學環境學院,北京 100084; 2.中國科學院生態環境研究中心,北京 100085)
8.1環境催化及其特殊性
自從1836年由瑞典化學家Berzelius提出催化(Catalysis)概念以來,催化科學和技術取得了長足進步,成為現代工業文明得以實現的重要基石之一。事實上,催化技術是化學工業和石油化學工業最核心技術。例如,80%以上的化學工業涉及催化技術,全球催化劑年銷售額超過100億美元,催化技術所帶來的產值達到其本身產值的百倍以上。發達國家國內生產總值的20%~30%來源于催化技術直接和間接的貢獻[1-2]。
掌握了催化科學和技術的人類在創造工業文明并給我們的物質生活帶來利益的同時,也使得人類對自然界的干預和改造能力大大加強,增加了人類活動結果超出環境所能承受范圍的風險。以下幾個例子清楚說明,催化是人類征服自然、改造自然的一把利劍,但運用不當也能成為危及人類可持續發展的雙刃劍。
20世紀初,Harber等開發出用于合成氨的鐵催化劑,實現了從氮氣和氫氣直接合成氨,從而造就了現代化肥工業,大大提高了農業產量,養活了地球上超過60億的人口,這項研究獲得了1918年諾貝爾化學獎。然而,正是由于現代農業大量施用氮肥造成了普遍的水體富營養化,同時人口劇增也挑戰了地球生態系統的負荷極限。
20世紀50年代初,Ziegler和Natta等發現了催化乙烯和丙烯聚合的Ziegler-Natta催化劑,并迅速實現了工業化生產,從此奠定了石化工業的基礎,這項研究獲得了1963年諾貝爾化學獎。然而,自然界中的微生物顯然無法降解人類在催化技術幫助下合成的高分子聚合物,大量合成和使用這類高分子聚合物造成了今天的“白色污染”。
自從掌握了以原油催化裂化和催化重整為代表的石油化工催化技術,人類得以從原油中獲得所需的汽油、柴油和煤油,從此交通運輸業得以迅速發展,人類迎來了通行便利的汽車時代。然而大量使用化石燃料消耗了地球寶貴的不可再生資源,并造成了嚴重的溫室氣體、酸雨和光化學煙霧等大氣污染,給人類的生存環境帶來巨大的壓力。
由此可見,催化科學和技術與人類今天面臨的環境和可持續發展問題關系密不可分。如今,已經取得巨大成功的催化科學與技術面臨著保護環境和顧及人類可持續發展問題的新挑戰。如果說催化技術是現代工業文明發展至今的基石之一,可以相信它也必將成為解決人類面臨的重大生存環境問題的關鍵技術,因此環境催化應運而生。
8.1.1環境催化的定義、研究對象和任務
對環境催化給出定義很困難,所以環境催化至今也沒有明確的定義。從催化化學的本質上看,所有的人為的催化過程和自然的催化過程都會對環境產生直接的或間接的影響。顯然,人為的所有催化過程中催化反應活性增加、選擇性提高和催化劑壽命增加都可以起到減少有害副產物、減少能源和原材料消耗、減輕環境負荷的作用,這些都可以為改善環境做出貢獻。反過來,掌握了催化技術的人類對自然界的干預和改造能力大大加強,這又增加了人類活動結果超出環境所能承受范圍的風險。從主觀上講,環境催化的概念是顧及人類可持續發展的環境友好的催化科學和技術。但從學科劃分來看,上述定義在目前看來顯然是過于寬泛,和現有的知識體系和學科結構有所沖突。不僅如此,這種依據主觀愿望所下的定義顯然不包括自然界自發的催化過程,然而這種不以人的意志為轉移的催化過程都會對環境產生這樣或那樣的作用。根據作者理解,環境催化應該包括人為的環境催化和自然界中自發的環境催化[3-4]。人為的環境催化內容僅限于在以下過程中所研究和使用的催化科學和技術:(1) 消除已經產生的污染物(環境催化的狹義定義);(2) 減少能源轉化過程中有害物質的產生(例如天然氣催化燃燒,柴油催化脫硫等);(3) 將廢物轉化為有用之物(例如二氧化碳的資源化)。自發的環境催化可以將整個地球大氣層看成一個光和熱的反應器,僅限于研究和地球表面以及大氣顆粒物有關的非均相大氣化學中的界面催化過程。應當指出,是否應該將自然界自發的催化過程歸屬到環境催化的范疇,研究者沒有形成統一的意見[3-6]。從廣義上講,凡是涉及到可以減少污染物排放的綠色催化過程都可以屬于環境催化的范疇,如化學計量催化技術(催化分子氧烯烴環氧化)、手性催化技術、替代有毒有害化學品的催化技術(氟氯烴替代)、產生清潔能源(光催化分解水)的催化技術等。
根據以上對環境催化的定義,本章中環境催化的研究對象和任務是,通過催化科學和技術的研究和應用,消除已經產生的污染物;減少能源轉化過程中有害物質的產生;將廢物轉化為有用之物;闡明非均相大氣化學中自發的界面催化過程,以增進了解污染物在環境微界面過程中的遷移轉化規律。環境催化的任務主要分為以下幾個方面:
8.1.1.1消除已經產生的污染
(1) 消除大氣污染物、溫室效應氣體和臭氧層消耗物質。大氣中主要氣態污染物有氮氧化物(NOx)、二氧化硫(SO2)、一氧化碳(CO)、二氧化碳(CO2),甲烷(CH4)、氟氯化烴(CFC)、非甲烷揮發性有機物(VOC)及羰基硫(COS)等。NOx和SO2對人體有害,經過大氣氧化過程后可以導致干、濕沉降(酸雨),其中NOx還可以和VOC發生復合污染導致光化學煙霧和近地層臭氧濃度升高。CO2、CH4和N2O是主要的溫室效應氣體,導致大氣層升溫;其中長壽命的N2O上升到臭氧層后被氧化成硝酸鹽,進而成為臭氧分解的催化劑。CFC和COS壽命也很長,上升到臭氧層后成為主要的臭氧消耗物質。除CFC外,上述氣態污染物都可以經由天然源的自然過程排放,氣態污染物的工業排放源又可以分為移動源(機動車)排放和固定源(發電廠、鍋爐、垃圾焚燒等)排放。
移動源排放的催化凈化方面,目前的三效催化劑(TWC)可同時去除汽油車尾氣中碳氫化合物(HC)、CO和NOx。固定源排放的催化凈化方面,根據火力發電廠、工業鍋爐、垃圾焚燒等固定源的排放特點,成熟的技術主要是以氨為還原劑的選擇性催化還原NOx(NH3-SCR)。溫室效應氣體和臭氧層消耗物質的催化轉化,目前的研究主要集中于可導致溫室效應的四種長壽命氣體的多相催化轉化。
(2) 消除室內氣態污染物和致病微生物。針對室內空氣化學污染和微生物污染,目前的研究主要集中于光催化凈化、催化氧化凈化和催化空氣滅菌(抑菌)。
(3) 消除水中污染物和致病微生物。目前的研究主要集中在飲用水和廢水處理過程中的催化應用。
8.1.1.2減少能源轉化過程中有害物質的產生
化石燃料燃燒過程中排放的CO2、SO2和NOx造成了大氣污染,產生溫室效應、酸雨、顆粒物和光化學煙霧等。針對移動燃燒源和固定燃燒源排放NOx、SO2、揮發性有機化合物和天然氣的催化凈化主要是多相催化燃燒。
8.1.1.3將廢物轉化為有用之物
利用豐富、廉價的有機廢棄物如纖維素等生物質資源生產燃料乙醇,有望替代傳統的化石燃料,從而實現能源的再生和可持續發展。最近研究表明[7-8],與傳統纖維素降解方法相比,催化氫解纖維素實現了纖維素降解為多元醇的綠色過程。這些剛剛起步的研究今后很可能為生物質資源轉化和資源化利用提供關鍵技術和解決方案。
8.1.1.4非均相大氣化學中的催化過程
前面所介紹的環境催化都是在人的主觀愿望指導進行的,然而自然界中還存在著不以人的意志為轉移的環境界面過程,即自發的環境催化過程。這里可將整個大氣層看成一個光和熱的催化反應器,在自然條件下,地表及大氣層中顆粒物表面上自發的多相催化反應對氣態污染物在自然界的遷移轉化產生影響,這種影響進而可以波及到相關元素的循環和整個大氣化學過程。討論的對象包括對流層中的地殼元素氧化物、無機鹽和冰顆粒物,研究的內容是常見的污染物在顆粒物表面的吸附、表面反應、脫附行為。
放眼21世紀,以消除環境污染物質、減輕環境負荷、將廢物轉化成有用之物為目的的環境催化已經日益成為催化工業的主流。根據催化劑集團網站數據顯示[9-10],1999年世界全球催化劑市場銷量為86億美元,其中環境催化劑銷量為26億美元,所占比例為29%,占據世界催化劑市場最大份額。到2006年,全球催化劑工業價值約為140~145億美元。其中環境催化劑部分銷售額約為52億美元,所占比例約為37%,是全球催化劑市場增長最快的部分,年增長率7%~9%。
8.1.2環境催化的特殊性
根據環境催化的定義,環境催化無論是研究對象還是工作條件都和通常的工業催化有很多區別。表8-1從反應物濃度、反應毒物濃度以及反應條件三個方面總結了環境催化和工業催化的區別[11-15]。

表8-1 環境催化與工業催化的區別
通常,工業催化所面對的反應物都會經過一定程度的精制,盡可能地去除對反應有害的物質。反應條件方面,可以根據催化反應的特點將反應溫度、壓力和空速設定在最大限度發揮催化劑作用的范圍。與此相對的環境催化,所面對的反應物濃度通常在10-6甚至于10-9級,這種稀薄的程度顯然無法進行任何濃縮和精制。同時,對環境催化有害的物質卻常常是反應物的數百倍甚至數萬倍,并且根本無法去除和避免。環境催化經常需要面對很高的空速、無法調整的溫度條件以及劇烈變動的反應負荷。例如,柴油機尾氣催化凈化過程要求催化劑能夠在將近10%氧氣氣氛中、有SO2和顆粒物等毒物共存時、在較低的溫度和較高的空速下、利用有限的還原劑選擇性地將只有10-3~10-4體積比濃度的NOx還原成氮氣,并且這個過程中催化轉化器還必須承受反應條件的劇烈變化。對于自然界自發發生的環境催化劑過程,研究者還必須面對一個多組分、多介質、復雜過程的自然環境[16-18]。
長期以來工業催化的許多成功是在大量反復試驗中取得,很大程度上依靠研究者的直覺和經驗。而這種研究模式越來越難以滿足目前環境催化過程中對催化劑活性、選擇性和穩定性更高的苛刻要求。面對如此苛刻的環境催化條件,出路在于能夠在理論的指導下設計出高低溫活性和高選擇性的催化劑,而這必然要求研究者對環境多相催化微觀過程如反應機理和催化活性中心結構有深入了解。多相催化是一個表面物理和化學過程,對這一表面過程的分子水平上的理解必定會極大地幫助我們最終設計催化劑[19-20]。
8.2移動源燃燒排放的催化凈化
機動車如同一把雙刃劍,在帶給人類便捷與舒適的同時也對大氣環境造成了巨大污染。隨著機動車數量的快速增長,機動車排放的污染物在城市大氣污染中的分擔率越來越高。按所使用的燃料區分,機動車可分為汽油車、柴油車和代用燃料車。機動車排放的尾氣成分中,除氮氣、氧氣、以及燃燒產物二氧化碳和水為無害成分外,其余均為有害成分。對于汽油車,HC、CO和NOx是三種主要污染物,而柴油車的主要污染物是顆粒物(PM)和NOx。其中大量排放的CO與人體血紅蛋白結合會造成輸氧功能下降,有些未燃燒完全的HC是致癌物質,會引發肺癌和甲狀腺癌等疾病,機動車排放尾氣中的細小PM會導致空氣能見度下降,給人體的呼吸系統帶來傷害。大量NOx的排放帶來了更嚴重的環境污染問題,NOx可以導致酸雨形成、水體富營養化、大氣能見度下降和光化學煙霧反應發生,在人體健康方面,NOx會降低人體的肺功能,破壞呼吸道的自然凈化機能,增加過濾性毒菌感染的易感性,降低人體對病毒感染的抵抗力。因此,有效控制機動車尾氣污染物的排放和消除機動車尾氣污染物具有重要的實際意義。
8.2.1汽油車尾氣催化凈化
西方國家從20世紀60年代開始進行機動車尾氣催化凈化的研究,70年代中期開始安裝含有Pt-Pd的氧化型催化劑[21],主要目的是控制CO和HC排放,并通過EGR方法減少NOx的排放。1976年聯邦德國的Robert Bosch GmbH成功研制了能夠嚴格控制機動車空燃比的氧傳感器,同年該項技術在Volvo和Saab汽油車上首次得到應用,1980年機動車用氧傳感器進入美國市場,1993年歐洲大部分國家要求所有的汽油車安裝氧傳感器[22]。自此,能同時催化凈化CO、HC和NOx的三效Pt-Rh貴金屬催化劑開始在汽油車制造業大規模使用。
8.2.1.1汽油車尾氣排放特點
汽油車尾氣中的主要污染物為CO、HC和NOx,而對于柴油車,CO和HC的排放相對于汽油車有所降低外,NOx和顆粒物成為主要尾氣排放污染物。表8-2給出了典型汽柴油發動機的尾氣排放情況。

表8-2 汽柴油發動機的尾氣排放情況[23]
①我國汽柴油的含硫量較高,實際數值應大于表中的數據
目前,汽油車排氣后處理技術的核心是三效催化技術。然而,三效催化轉化器的工作狀態與發動機的空燃比密切相關,三效催化轉化器必須在一定的空燃比范圍,即在三效催化劑工作窗口才能正常工作。圖8-1給出不同空燃比條件下三效催化轉化器對主要污染物HC、CO和NOx的催化凈化效果。從圖8-1可以看出,只有發動機在理論空燃比附近工作,三效催化劑才能同時將汽車尾氣中的主要污染物CO、HC和NOx轉化為無害的CO2、H2O和N2。富燃條件下由于氧氣不充足,使CO和HC的轉換率下降,在稀燃條件下CO和HC可以完全氧化,而NOx很難被完全還原。

圖8-1 三效催化劑的工作窗口
8.2.1.2催化轉化器
催化轉化器是由殼體、減振層和催化劑構成。其中催化劑是指載體、涂層和催化活性組分,是整個催化轉化器的核心部分,決定著催化轉化器的主要性能指標。最早的催化轉化器中的催化劑是以球狀氧化鋁(γ-Al2O3)為載體,穩定劑和活性組分涂覆在表面,然后填裝在殼體內,這種載體存在磨損快,阻力大的缺點。后來發展成為蜂窩狀的堇青石陶瓷或不銹鋼載體上負載涂層和活性組分的整體式催化劑,如圖8-2和圖8-3所示。催化轉化器在催化劑外面包裹減振層,最后由不銹鋼殼體封裝而成。

圖8-2 催化轉化器的基本結構

圖8-3 三效催化劑的結構和涂層
殼體:催化轉化器殼體一般為不銹鋼板材,以防止因氧化殼體脫落造成催化劑堵塞。許多催化轉化器殼體做成雙層結構以保證催化劑的反應溫度。
減振層:減振層一般有膨脹墊片和鋼絲網墊兩種,起減振、緩解熱應力、固定載體、保溫和密封作用。膨脹墊片由蛭石(45%~60%)、硅酸鋁纖維(30%~45%)以及黏接劑組成。膨脹墊片第一次受熱時體積明顯膨脹,而冷卻時僅部分收縮,使金屬殼體與陶瓷載體之間的縫隙完全脹死并密封。
催化劑載體:陶瓷蜂窩載體最早由美國康寧(Corning)公司生產,隨后日本NGK公司也掌握了此技術,并開始大量生產。陶瓷蜂窩載體材料為多孔堇青石(2MgO·2Al2O3·5SiO2)陶瓷,化學組成為ω(MgO)=14%、ω(Al2O3)=36%和ω(SiO2)=50%。目前全球汽車用催化器載體90%是陶瓷載體,也有一部分車型的三效催化劑使用金屬蜂窩載體,如Audi和Volvo等品牌的某些車型。金屬蜂窩載體與陶瓷蜂窩載體相比具有導熱率高、開孔面積大、孔壁薄和機械強度高等特點,對汽油車冷啟動階段的污染排放控制和延長三效催化劑的使用壽命大有裨益。此外,摩托車由于振動顛簸原因,排氣污染控制催化劑的載體也多采用金屬載體。
8.2.1.3汽油車排放污染控制三效催化劑的研究
現狀和發展
從20世紀70年代開始,汽油車排氣污染控制技術伴隨著發動機和車輛制造與控制技術的進步以及汽車排放法規的日益嚴格而逐步完善。可以追蹤的足跡為[24]:

綜上所述,在過去的30年中,汽油車尾氣排放控制三效催化劑技術得到長足發展,利用先進的尾氣排放控制技術、先進的發動機燃燒控制和汽車制造技術,可以制備出超低排放或超超低排放的整車。但三效催化劑制造技術還是不斷地受到日益嚴格的排放法規、日益枯竭的貴金屬資源和日益增長的貴金屬價格的挑戰。目前,三效催化劑的研究與開發重點是在滿足日益嚴格的排放法規基礎上,降低貴金屬用量或尋找部分取代貴金屬的技術路線,而納米三效催化劑和稀燃發動機排氣控制技術是這一領域研究的核心。
(1) 穩定氧化鋁的研究。如圖8-3所示,三效催化劑載體分為蜂窩陶瓷(或金屬)載體和γ-Al2O3涂層(washcoat)兩部分。蜂窩陶瓷(或金屬載體)的幾何表面積約為(2.0~4.0) m2·L-1,如此小的表面積不足以提高負載貴金屬的表面空間,因此需要在其上涂覆一層氧化物作為三效催化劑的第二載體,通常稱之為“水洗涂層”(washcoat),以擴大催化劑載體的比表面積,俗稱為“擴表”。由于γ-Al2O3具有高的比表面積和高溫水熱穩定性,γ-Al2O3通常被選擇為三效催化劑的第二載體。α-Al2O3的高溫熱穩定性高于γ-Al2O3,但比表面積通常小于10 m2·g-1,經常用作高溫條件下的催化劑載體。例如,汽油車的密偶催化劑安裝在發動機歧管出口處,需要經受1 000℃以上的高溫,因此,密偶催化劑經常采用α-Al2O3作載體[23]。
(2) 三效催化劑的活性組分。無可置疑,貴金屬元素Pt、Rh和Pd在三效催化劑中起關鍵作用,三效催化反應在Pt、Rh和Pd原子組成的原子簇或活性中心進行。理解這三種貴金屬在催化性質上的異同對開發三效催化劑十分重要。
Pt在三效催化劑中主要是催化CO和HC的完全氧化反應。在早期采用的雙段催化床催化轉化器中,后段床氧化型催化劑主要成分是Pt。Pt對NO有一定的還原能力,但當尾氣中CO濃度較高或有SO2存在時,Pt對NO的凈化效果比Rh差,并且Pt還原NOx的窗口較窄,在還原型氣氛中容易將NOx還原為氨氣。Pt在三效催化劑中的典型用量為(1.5~2.5) g·L-1。據1990年統計,用于汽車催化劑的Pt約占西方總市場消費量的36%。
Rh是三效催化劑中控制NOx的主要活性成分,可在較低的溫度下選擇性還原NOx為氮氣,同時產生少量的氨。在實際的尾氣反應中,還原劑可以是CO、HC,還可以是H2。氧氣對NOx還原反應影響很大,在有氧條件下,N2是惟一的還原產物,在無氧條件下,低溫下的主要還原產物是NH3,高溫下的主要產物是N2。此外,Rh對CO的氧化以及HC化合物的重整反應也有重要的催化作用,與Pt和Pd催化劑相比,Rh催化劑對于CO和HC的催化活性較低。但無論如何,Rh在三效催化劑中不可或缺,沒有Rh的存在,NOx含量往往不能達到排放標準。
Pd催化劑在一定條件下可以具有很好的三效催化活性,早在1975-1976年,Pd就被用來制造汽車尾氣污染排放控制催化劑,到1990年代中期,Pd的三效催化反應活性得到深入研究,形成了單Pd三效催化劑制備技術[25]。該技術采用分層負載Pd和CeO2以及堿土金屬氧化物,使單Pd催化劑具有很好的三效催化活性,單Pd三效催化劑的結構如圖8-4所示。在Pt、Rh和Pd貴金屬元素中,Rh無疑是最重要的一個,可以促進NO的解離,提高NO去除效率[26-27],在三效催化劑中不可缺少。
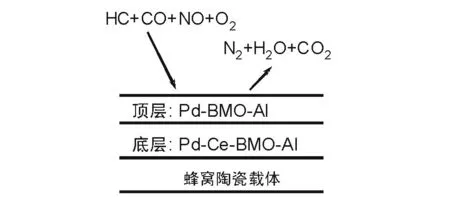
圖8-4 單Pd催化劑的層狀結構
(3) 三效催化劑中的儲氧材料。在汽車尾氣催化劑中使用CeO2或CeZrO2材料可以追溯到20世紀80年代[28],催化劑主要活性組分已經包括Rh和Pt貴金屬以及作為儲氧材料的CeO2。汽車尾氣中污染物的高效凈化需要在化學計量比即在理論空燃比(λ=1或A/F=14.7)條件下進行,遠離理論空然比時,三效催化劑的效率大大降低。因為汽車發動機的排氣特性是以一定頻率,一定振幅以理論空燃比為中心振蕩,在遠離理論空燃比時,三效催化劑的催化效率受到極大限制。氧化還原反應Ce4+?Ce3+賦予了CeO2材料儲放氧的功能,即在富燃工況下,CeO2釋放出O2,稀燃工況下,又吸收和儲存O2,從而達到調節汽車尾氣中氧含量的目的。最早的儲氧材料是單純的CeO2,而目前使用的儲氧材料大多為CeO2-ZrO2固溶體。研究發現,CeO2或CeO2-ZrO2固溶體不僅具有儲存氧的功能,還對三效催化劑的性質有更多重要的影響。
8.2.2柴油機和稀燃汽油機尾氣催化凈化
傳統的汽油機在理論空燃比(14.7)附近的狹窄范圍工作,一定程度上犧牲了燃油的經濟性。隨著對全球能源危機和溫室效應加劇的關注,對降低CO2排放和節約能源提出了更高要求。在這種背景下,稀燃技術引起廣泛關注。稀燃汽油機燃燒經濟性好,污染物排放量低。柴油機也是典型的稀燃發動機,自1892年問世以來,憑借其良好的動力性、經濟性和耐久性等優點在車用動力中占據重要位置,隨著全球石油資源短缺的加劇,其重要性愈發明顯。自20世紀70年代,歐洲和日本就基本實現了載貨汽車和大型客車的柴油機化。目前,歐洲轎車年產量中40%已采用柴油發動機,在法國、西班牙等更高達50%以上。
與汽油機相比,稀薄燃燒發動機是一種環境友好的發動機,采用富氧燃燒技術抑制了CO和HC的形成,但與裝配了三效催化劑的汽油車相比,柴油機的氮氧化物(NOx)和顆粒物(PM)排放和稀燃汽油機的NOx排放成為制約其推廣的重要因素[29]。柴油機尾氣的兩大污染物NOx與PM的形成與含量存在相互制約關系,圖8-5清楚地表明了這一點:努力減少其一,必然導致另一污染物增加,即通過機內措施同時減少或消除NOx和PM的排放極其困難;另外,盡管機內凈化技術使顆粒物的排放總量得以削減,卻生成了對人體危害更大的微細顆粒物,而未來的法規將會對柴油機顆粒物排放的數量進行限制。柴油車尾氣凈化技術主要包括NOx催化凈化、氧化催化技術(DOC)和柴油車顆粒物過濾器技術(DPF)。針對柴油機和稀燃汽油機尾氣NOx的催化凈化,目前主要研究方向有貯存-還原氮氧化物(NSR)和選擇性催化還原氮氧化物(SCR)。

圖8-5 重型柴油機尾氣NOx和PM的Trade-off關系與排放標準[30]
8.2.2.1選擇性催化還原氮氧化物技術(SCR)
富氧條件下NOx選擇性催化還原是指在催化劑的作用下,通過抑制還原劑的非選擇性氧化,從而促進還原劑與NOx反應形成N2的過程。由于柴油機采用富氧燃燒技術,導致尾氣中未燃HC的絕對量不足,需要另行添加還原劑以凈化NOx。根據外加還原劑的不同,可分為氨類(尿素)選擇性催化還原NOx與碳氫化合物選擇性催化還原NOx。
(1) 尿素選擇性催化還原氮氧化物(Urea-SCR)。針對固定源尾氣例如燃煤電廠煙氣和固定型柴油機尾氣中NOx的去除,可以采用氨選擇性催化還原NOx技術(NH3-SCR),即使用氨水或液氨作為還原劑選擇性還原NOx。目前該技術已在國外廣泛應用于固定源煙氣脫硝,其原理是利用V2O5/TiO2催化劑,在O2大大過量的條件下,讓NH3選擇性還原NOx到N2。使用以TiO2為基礎的催化劑,保證了催化轉化器對SO2有很強的耐受性。目前,NH3-SCR已經應用于重型柴油機尾氣NOx凈化。針對移動源特別是柴油車的特點,氨水或液氨在儲存和運輸上存在危險性,且對存儲設備具有腐蝕性,因此尿素可替代NH3作為柴油車用SCR還原劑。尿素是白色顆粒或結晶狀的固體,理論含氮量46.65%,是無毒、不具有腐蝕性的物質。一分子Urea水解可生成兩分子NH3和一分子CO2,因此被視為NH3的有效儲存源。Urea-SCR體系中使用Urea質量分數為32.5%的水溶液,稱為“AdBlue”。使用Urea作為還原劑可以克服因使用氨水或液氨造成的諸多問題,因此Urea-SCR也成為目前重型柴油機尾氣去除NOx的首選技術。
目前工業Urea-SCR催化劑體系和固定源煙氣脫硝所用催化劑體系基本相同,主要是V2O5-WO3/TiO2或V2O5-MoO3/TiO2體系。其原理是在O2大大過量的條件下讓Urea水解生成的NH3選擇性還原NO到N2。在較寬的溫度范圍Urea具備了優異的選擇性還原NOx的能力,該催化體系在(260~500)℃凈化率可達90%以上。
一般而言,V2O5-TiO2體系應用于選擇性催化還原固定源尾氣NOx的空速為10 000 h-1,而三效催化劑可在空速高達100 000 h-1時高效工作。因此,為高效去除柴油機尾氣中的NOx,需要大體積催化劑。如SINOx只用SCR催化劑來凈化卡車尾氣中的NOx,對氣缸體積為12 L的發動機,需要30 L 200目的V2O5-WO3/TiO2整體催化劑才能獲得滿意的效果[31]。
用Urea還原NOx時須將該還原劑先水解成NH3,然后以NH3的形式參與隨后的反應,同樣會面臨NH3的泄漏問題,尤其是在發動機工況瞬間變化導致排氣溫度升高時,增加了控制的難度[31]。為了提高Urea-SCR體系凈化NO的效率,減少NH3的泄漏,MAN公司提出了基于Urea-SCR體系的VHRO系統,如圖8-6所示。“V”為前置的氧化催化劑,該催化劑的作用是將排氣中的部分NO氧化為NO2,以提高SCR催化劑的低溫活性;“H”為Urea水解催化劑,其作用在于加速Urea水解,從而有利于隨后的NOx選擇性還原;“R”為SCR催化劑,在該催化劑床層中,排氣中的NOx被Urea水解形成的NH3選擇性還原為N2;“O”為NH3選擇性氧化催化劑,可將排氣中剩余的NH3轉化為N2,減少NH3的泄漏。

圖8-6 MAN公司提出的“VHRO”體系[32]
Urea-SCR技術中,傳統工業催化劑(V2O5-WO3/TiO2或V2O5-MoO3/TiO2)雖然具有催化活性高、抗SO2中毒性能好的優異性能,但仍存在著:① 催化劑體系中含有有毒物質V,使用過程中容易發生脫落,對生態環境和人體健康存在危害;② 催化劑操作溫度高且窗口較窄;③ 使用溫度較高時(超過400℃),N2O生成量較大,SCR反應的選擇性降低。因此越來越多的研究者開始致力于開發新型的Urea-SCR催化劑體系,以期解決上述問題。
隨著柴油機尾氣凈化技術發展,顆粒物捕集器(DPF)將置于SCR催化體系之前,以實現顆粒物和NOx的同時削減。DPF再生時引發的高溫水熱環境,對后置的SCR催化劑提出了更高要求。從目前研究和應用情況看,新型Urea-SCR催化劑體系主要包括金屬氧化物和分子篩催化劑兩類。
研究較多的非釩金屬氧化物催化劑主要有Fe基、Ce基和Mn基等氧化物催化劑,如鐵鈦復合氧化物(FeTiOx)[33-34]、鈰鈦復合氧化物CeTiOx[35]以及具有優異抗高空速性能的鈰鎢復合氧化物CeWOx[36]。雖然非釩金屬氧化物催化劑種類繁多,但通常存在高溫穩定性和抗硫抗水性能不足等問題,目前尚未得到實際應用。
據報道,許多金屬分子篩催化劑均在NH3-SCR反應中有較高的活性,如Cu、Fe、Mn和Ce等。在這些催化劑中,Cu基和Fe基分子篩催化劑是最為活潑的SCR催化劑,并得到廣泛研究。特別是Cu-ZSM-5、Fe-ZSM-5等催化劑,表現出較高的NH3-SCR反應活性、較好的抗水抗硫性能和穩定性,被看作是最有可能在機動車上廣泛應用的催化劑之一。Fe基分子篩具有優越的水熱穩定性,但其優勢活性溫度窗口在350℃以上,低溫活性較差。Cu-ZSM-5等具有優良的低溫活性,但水熱穩定性較差。近年來,CHA結構的Cu基小孔分子篩Cu-SSZ-13和Cu-SAPO-34由于具備優良活性和高水熱穩定性引起廣泛關注[37-38]。
福特汽車公司[39]經過長時間水熱老化Fe基和Cu基分子篩催化劑,然后將其活性和應用于固定源的傳統V基催化劑進行對比(圖8-7)。該實驗選用水熱老化條件為670℃老化64 h,這代表裝有DPF的柴油車上的SCR催化劑運行120 000英里。結果發現,V基SCR催化劑顯然不能用于美國柴油車尾氣凈化,而Fe基和Cu基催化劑是較好的選擇。Cu/分子篩和Fe/分子篩具有良好的水熱穩定性,和Cu/分子篩相比,而Fe/分子篩催化劑低溫活性不佳,同時具有較高的高溫活性。
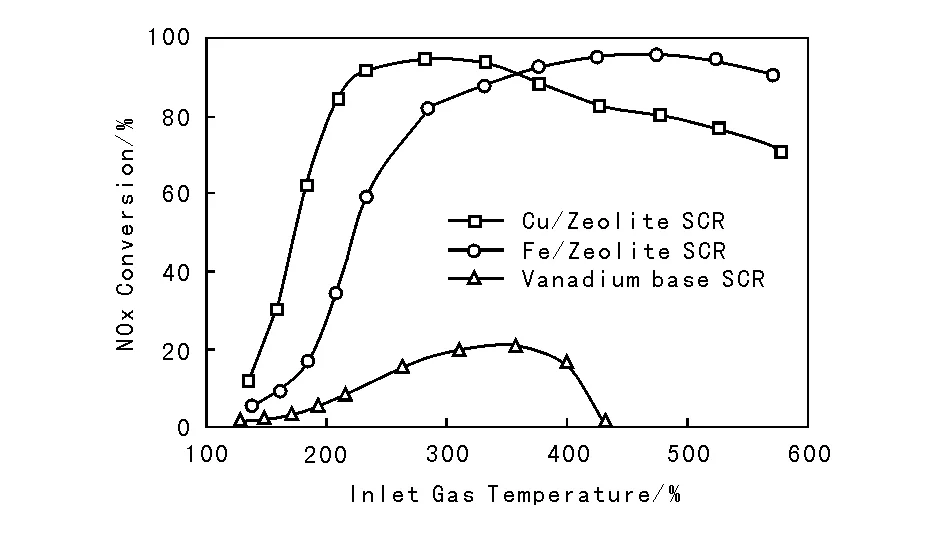
圖8-7 Cu、Fe和V基蜂窩狀催化劑NH3-SCR反應的NOx轉化率[39]
(2) 碳氫化合物選擇性催化還原氮氧化物(HC-SCR)。20世紀90年代初,Sato S等[40]率先報道了富氧條件下,Cu-ZSM-5具有較好的催化低碳氫化合物選擇性還原NOx活性。隨后碳氫化合物選擇性催化還原NOx(HC-SCR)引起研究者的廣泛興趣。研究表明,分子篩催化劑、負載型的貴金屬催化劑、金屬氧化物催化劑均具備催化碳氫化合物選擇性還原NOx的能力。其中,Ag/Al2O3催化劑具有優異的催化碳氫化合物及含氧碳氫化合物選擇性還原NOx的活性,是最有望實用化的催化劑之一[41-43]。
與尿素-SCR相比,HC-SCR技術的優勢在于能以車載燃油作為還原劑的來源,因而可以免除還原劑添加基礎設施的建設,簡化后處理系統。但直接以柴油為還原劑時,NOx的凈化效率與耐久性還難以滿足排放法規的要求[44]。與此相反,乙醇等小分子含氧碳氫具備了優異的選擇性催化還原NOx效率[41-43]。鑒于此,Yu Y B等對Ag/Al2O3-乙醇這一高效的催化劑-還原組合體系進行了深入研究,利用原位紅外技術并輔以量化模擬,首次在Ag/Al2O3催化乙醇部分氧化及其選擇性催化還原NOx過程中發現了表面烯醇式物種(enolic speces)[45]。Ag/Al2O3催化劑表面吸附態的烯醇式物種能與NOx迅速反應,轉化為最終產物N2,從而揭示了乙醇選擇性還原NOx高效特性的微觀機制[42,46]。隨后的研究表明,烯醇是碳氫化合物及含氧碳氫化合物氧化過程中關鍵的中間體[47]。

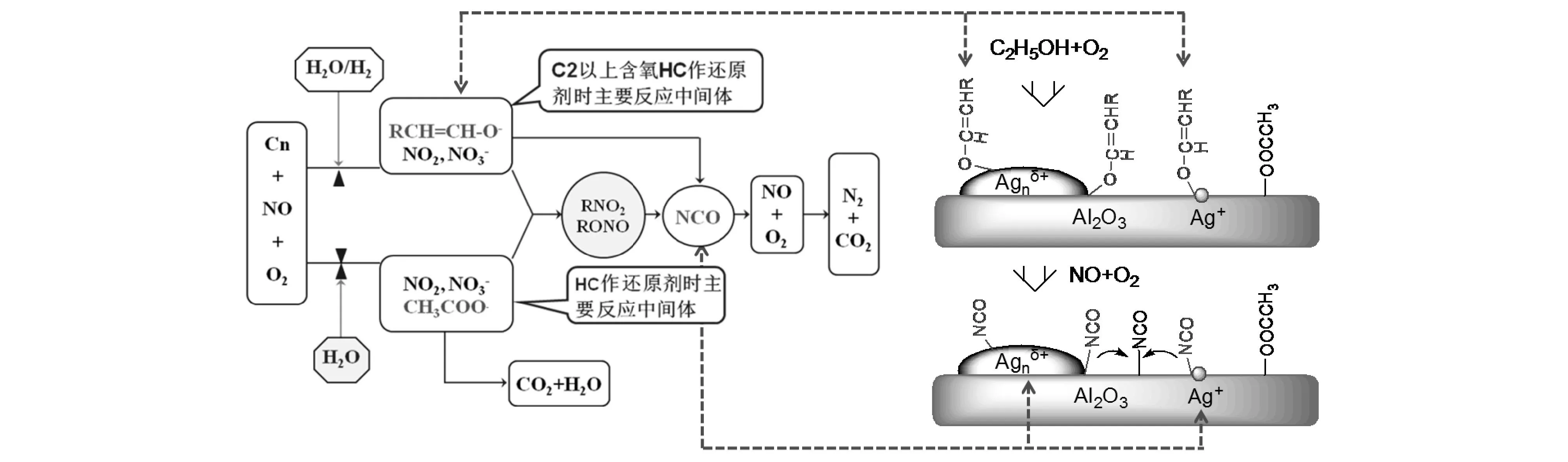
圖8-8 銀/氧化鋁催化劑上HC-SCR反應機制與構效關系
8.2.2.2貯存-還原氮氧化物(NSR)
NOx貯存-還原(NSR)技術首先以稀燃汽油機尾氣處理為對象研究開發。因為柴油車和稀燃汽油車的排放尾氣都為富氧氣氛,所以NSR催化劑在解決了硫酸鹽中毒的條件下也可以處理柴油車尾氣。20世紀90年代中期,在研制和評價汽油車三效催化劑的過程中,日本豐田汽車公司的技術人員發現一種現象:在大于理論空燃比(λ>1)的尾氣條件下,排放尾氣中的NOx可以被一種含有貴金屬和堿土金屬氧化物的三效催化劑大量吸附。
大部分NSR催化劑配方是以貴金屬作為催化活性組分,以堿和/或堿土金屬氧化物作為NOx儲存材料,其反應機理示意圖如圖8-9所示[50]。隨著研究深入,日本豐田汽車公司已將NSR技術與顆粒物捕集技術成功整合,在日本輕型柴油車上進行了示范應用,并大力向歐洲推行該技術。

圖8-9 吸附儲存還原NO的反應機理[50]
8.2.3清潔燃料車尾氣催化凈化
能源短缺和城市群復合大氣污染成為全球性問題,選擇低排放、資源豐富易得的新型燃料替代汽油已經成為一種趨勢。目前,替代燃料研究主要包括壓縮天然氣(CNG)、液化石油氣(LPG)、含氧燃料如醇類、醚類等,此外還有生物柴油、氫氣、電等。
我國天然氣資源豐富,預測儲藏量為38萬億立方米,而且氣質良好,甲烷含量90%以上,含硫少。使用天然氣作為汽車燃料,可以大大降低發動機廢氣排放中的主要有害成分。其中未燃燒甲烷等成分性質穩定,在大氣中不會形成有害的光化學煙霧;同時,天然氣汽車的使用成本較低,比燃油汽車節約燃料費約50%;而且與電動汽車相比,天然氣汽車的續駛里程長,因此,天然氣汽車是目前被認為最具有推廣價值的低污染汽車之一,尤其適合于城市公共交通和出租汽車使用[51-52]。目前中噸位的商用車用壓縮天然氣的開發目標是能顯著降低氮氧化物的排放并保持與柴油型發動機相同的動力性能。然而,天然氣汽車降低CO和碳氫排放的同時,甲烷的排放量增加。甲烷作為一種溫室氣體,對大氣的增溫潛勢是CO2的32倍,對溫室效應的影響更為嚴重,需要進行有效控制,此外如何去除CNG汽車尾氣中的氮氧化物仍然是一個難題。
含氧燃料車中使用最多的是乙醇,此外還有甲醇、二甲醚等。乙醇可以從生物質制取,以乙醇為燃料的發動機所排出的CO2被植物所吸收,乙醇成為一種能夠滿足可持續發展要求的燃料[53-54]。乙醇汽油在巴西和美國等國家已經廣泛應用,乙醇汽油中的乙醇濃度為10%~85%。在美國有500萬輛具備燃燒含85%乙醇汽油的車已經上路,巴西全國標準燃料是25%的乙醇燃料,乙醇燃料的含量范圍可擴至100%乙醇。我國已經在許多省市推廣應用含10%的乙醇-汽油混合燃料,以改善汽車能源結構,并于2001年頒布了變性燃料乙醇國家標準(GB 18350-2001)和車用乙醇國家標準(GB18351-2001)。推廣應用乙醇汽油的同時,對發動機和催化轉換器也提出新的要求,含氧燃料中的氧含量會造成空燃比增大,氮氧化物不能有效去除。此外,乙醇汽油車不可避免地會排放一些醇類及部分氧化的醛和酸,這些化合物排放到大氣中對環境危害更大。因此,如何使三效催化劑能夠氧化CO、HC的同時繼續高效還原NOx并同時去除一些含氧有機物醇類及部分氧化的醛和酸,成為目前改進三效催化劑的一個研究課題。
8.2.3.1甲烷氧化催化劑
甲烷是最穩定的烴類,通常很難被活化或氧化,且甲烷催化燃燒工作溫度較高,燃燒反應過程中會產生大量水蒸汽,同時天然氣中含少量硫,因此,甲烷催化燃燒催化劑必須具備較高的活性和水熱穩定性以及一定的抗中毒能力。國內外研究者致力于研究開發高效穩定的甲烷低溫催化燃燒催化劑,主要包含貴金屬和氧化物催化劑兩類。
貴金屬是活性最高的燃燒催化材料,具有很高的低溫催化燃燒活性,并且在500℃以下具有良好的抗硫性能。但高溫穩定性較差,在1 000℃以上時,會因貴金屬粒子聚集、燒結、蒸發等失去活性。在眾多的貴金屬材料中,鉑和鈀的應用最為廣泛。貴金屬燃燒催化材料一般采用γ-Al2O3為載體,主要是利用其高比表面積、低成本特性。其他載體材料還可采用SiO2、SnO2、TiO2、CeO2-ZrO2、分子篩以及組合載體等。
金屬氧化物類催化劑一般具有結構穩定、耐高溫性能和抗中毒能力強以及高溫活性穩定等特點,目前主要用于甲烷的高溫燃燒。金屬氧化物與貴金屬相比在價格上具有一定優勢,但活性與貴金屬催化劑相比還有一定差距。金屬氧化物催化劑包括單組分金屬氧化物催化劑和復合金屬氧化物催化劑。單組分過渡金屬氧化物,如CuO、Co2O3、Mn2O3、Cr2O3等,都是良好的催化燃燒催化劑。一般來說,各種單金屬氧化物催化劑在甲烷燃燒中的催化活性依次為:Co3O4>CuO>NiO>Mn2O3>Cr2O3[55]。鈣鈦礦型(ABO3)復合氧化物催化劑、六鋁酸鹽及取代型六鋁酸鹽等的熱穩定性相對比簡單的金屬氧化物和貴金屬要好得多,被認為是高溫催化燃燒有廣闊應用前景的催化劑。
8.2.3.2甲烷選擇性催化還原氮氧化物催化劑
甲烷選擇性催化還原NOx(CH4-SCR)是利用甲烷作還原劑催化凈化NOx的有效方法,優點是CH4是天然氣中的主要成分,使得CH4較其他HC化合物更易獲得,且非常廉價。此外,相比NH3,CH4對設備的腐蝕性非常小,可降低設備投資。基于上述優勢,研究者開展了廣泛的CH4-SCR研究,并取得一定的進展。

8.2.3.3含氧燃料汽車尾氣凈化方法
在乙醇汽油車尾氣污染物中,HC和CO的排放可以明顯降低,但對減少NOx排放量的作用不大,甚至會增加NOx的排放量。一些非常規污染物乙醛、乙醇等低分子含氧有機物排放量也會增加。此外,乙醇汽油車尾氣容易使三效催化劑積炭,從而減少催化劑的使用壽命。為適應我國乙醇汽油的推廣應用,需要針對乙醇汽油車的排放特性開發出專用的催化劑,使其不但能夠從總體上控制CO、HC和NOx的排放,還能減少乙烯、乙醛以及芳香族化合物等有害物質的排放。
8.3固定源燃燒排放的催化凈化
固定污染源是指排放位置和地點固定不變的污染源,如電廠鍋爐、各種廠礦的工業鍋爐等。在固定污染源的燃料消耗中,燃煤占有相當大的比重。煤炭燃燒過程中會產生大量的污染物,排放的煙氣中對環境造成污染的物質主要是CO、SOx、NOx及可吸入顆粒物(PM)。對于CO,可以通過控制燃料在燃燒過程中的空燃比、燃燒溫度和燃燒時間,并使燃料和空氣混合均勻,從而使其燃燒完全,達到將CO濃度控制在排放標準以內的目的。但通過燃燒過程控制并不能將SO2和NOx完全消除,這些致酸物質的大量排放引起的酸沉降已經與臭氧層破壞、全球氣候變化一起成為最為突出的大氣環境熱點問題,影響范圍已經由局部性污染發展成為區域性污染,甚至成為全球性污染。同時,SO2和NOx的越境遷移問題也倍受關注。
8.3.1煙氣選擇性催化還原(SCR)脫硝原理和技術
選擇性催化還原法是目前應用最為廣泛的煙氣脫硝技術。該方法主要采用氨(NH3)作為還原劑,將NOx選擇性還原成N2。NH3具有較高的選擇性,在一定溫度范圍,它主要與NOx發生作用,而不被煙氣中的O2氧化,因而比無選擇性的還原劑脫硝效果好。當采用催化劑促進NH3和NOx的還原反應時,其反應溫度操作窗口取決于所選用催化劑種類,根據采用的催化劑不同,催化反應器應布置在局部煙道中相應溫度的位置。
8.3.1.1SCR的工作原理
對于固定源脫硝,主要是采用向溫度為(280~420)℃的煙氣中噴入尿素或氨,將NOx還原為N2和H2O。反應時,排放氣體中的NOx和注入的NH3幾乎是以物質的量比1∶1進行反應,可得到80%~90%的脫硝率[57]。NH3-SCR法去除NOx的基本原理如圖8-10所示。

圖8-10 SCR法脫硝基本原理
8.3.1.2SCR催化劑
在SCR技術應用過程中,催化劑的制備生產是其中最重要的部分之一,其催化性能直接影響SCR系統的整體脫硝效率。催化劑的更換與還原劑的消耗是SCR系統運行費用的最主要來源,同時催化劑的制備生產更是占據了SCR系統初期建設成本的20%以上。
(1) 高溫催化劑。高溫催化劑按照載體的區別,可分為分子篩和金屬氧化物催化劑,由于催化劑工作溫度在250℃以上,具有較好的抗水抗硫性能,因此具有一定的實際應用價值。① 分子篩類催化劑。分子篩類催化劑是研究非常活躍的一個領域,無論作為催化還原還是催化分解的催化劑,金屬離子交換分子篩都具有很高的活性。分子篩用作催化劑是基于其特殊的微孔結構,其類型、熱處理條件、硅鋁比、交換的離子種類、交換度等都會影響活性。目前已開展的研究中涉及了多種類型的分子篩,主要包括Y型、ZSM系列[58-59]、發光沸石(MOR)[60]和菱沸石(CHA)[37,61]等,而用于離子交換的金屬元素主要包括Mn、Cu、Co、Pd、V、Ir、Fe和Ce等。離子交換法制備的分子篩催化劑中,Cu-ZSM-5和Fe-ZSM-5催化劑因還原活性高、活性溫度區間寬而引起廣泛關注,研究者開展了大量研究,取得一些研究進展,并開始實際應用;② 金屬氧化物催化劑。金屬氧化物催化劑在富氧條件下的NH3選擇性催化還原NOx反應中表現出較好的催化活性,應用最多的是以V2O5為活性組分,將其負載于Al2O3、SiO2、Al2O3-SiO2、ZrO2、TiO2、TiO2-SiO2等氧化物上。
(2) 低溫催化劑。工業上應用最廣的SCR催化劑是V2O5-WO3/TiO2,該催化劑具有較高的脫硝活性和抗SO2、H2O中毒能力,但其催化劑成本和操作窗口均較高(>350℃)。為了避免煙氣的預熱耗能,降低脫NOx成本,研制開發與之匹配的低溫SCR催化劑成為熱點。① 貴金屬催化劑。貴金屬催化劑是研究較早的一類催化劑,通常以Pt、Rh和Pd等為活性組分,氧化鋁或整體式陶瓷為載體[62-63]。在這類催化劑中,較多采用CO以及碳氫化合物或CO、H2混合氣作為還原劑。貴金屬催化劑的應用研究目前還有待于進一步的實驗探索,低溫活性的進一步提高、抗硫性能的增強以及還原產物N2的選擇性問題都將是未來的主要研究目標;② 碳基載體的氧化物催化劑。活性炭以其特殊的孔結構和大比表面積成為一種優良的固體吸附劑,廣泛用于空氣或工業廢氣的凈化。實際上,在NOx的治理中,不僅可以作吸附劑,還可以作催化劑,在低溫(90~200)℃條件下,有NH3、CO或H2存在時可選擇性還原NOx;沒有催化劑時,還可以直接作還原劑,在400℃以上使NOx還原為N2,自身轉化為CO2。所以,活性炭在固定源NOx治理中有較高的應用價值,其最大優勢在于來源豐富,價格低廉,易于再生,適用于溫度較低的環境,這是使用其他催化劑所不能實現的。但活性炭作催化劑時活性很低,特別是空速較高的情況下。在實際應用中,常常需要經過預活化處理或負載一些活性組分以改善催化性能;③ 錳基氧化物催化劑。許多含有過渡金屬(Fe、V、Cr、Cu、Co和Mn)的SCR催化劑具有良好的低溫SCR活性,其中含Mn的SCR催化劑由于具有優越的低溫活性而得到廣泛研究,但其較差的抗H2O和SO2性能是該類催化劑實際應用的最大障礙。如果能通過改進配方,提高催化劑的抗H2O和SO2能力,則具有廣闊的應用前景;④ 分子篩載體催化劑。分子篩類型催化劑是研究非常活躍的一個領域,目前,分子篩載體也廣泛應用于低溫催化劑的研究中。
8.3.1.3SCR催化反應機理
(1) V2O5/TiO2等金屬氧化物催化劑上的反應機理。20世紀70年代以來,對V基催化劑上的SCR反應機理和潛在的活性位已進行了大量研究,這些研究建立在反應動力學和反應物吸附態光譜分析基礎上。研究發現,任何一種金屬氧化物,如果在催化氧化反應中有活性,對選擇性催化還原反應也同樣具有活性。催化劑組分中,如果以TiO2為載體,對于部分氧化具有高選擇性,那么同樣對SCR反應也具有高選擇性。過渡金屬氧化物對于氧化催化有低的活性,在SCR反應中的活性也較低。表明SCR反應是一個氧化還原反應,其機理是氧化還原機理,或Mars-van-Krevelen機理。


(8-1)

(8-2)
8.3.1.4SCR反應動力學
關于SCR反應化學動力學研究,大部分在接近“真實”反應條件下獲得。基本上同時采用兩種方法,即經驗方法和機理模型(如Langmiur-Hinshelwood或Eley-Rideal模型)。一般認為NO的轉化速率與反應物NO、NH3、O2的濃度(cNO,cNH3,cO2)有關,其動力學關系式一般表示為:
γNO=kccNOαcNH3βcO2γcH2Oδ
(8-3)
其中,NO濃度反應級數近似認為1[65-68]。也有一些研究測出更低的α值(0.5~0.8)[69]。Odenbrand C U I[70]和Boulahouache A等[71]發現α值隨反應溫度升高而增大,對于Cr2O3/TiO2,Fe-Y,Cu-ZSM-5和其他Cu-Exchanged沸石催化劑,NO的反應級數也近似為1。另外,Komatsu T等[72]測得在Fe-ZSM-5催化劑上α=0.8;Willey R J等[73]測得在鐵的氧化物上反應時α=0.64。
在富氧和水蒸汽含量大于5%時,上式中CO2和H2O濃度可以忽略,在這種條件下并且當NH3/NO>1時,根據Hansen A C等[53]對V2O5/TiO2的研究,反應式可寫為:
γNO=kccNOα
(8-4)
這意味著β為0。
8.3.1.5SCR系統及應用
選擇性催化還原脫硝系統主要包括脫氮反應器、還原劑儲存及供應系統、氨噴射器和控制系統四個部分(見圖8-11)。

圖8-11 SCR脫硝系統示意圖
(1) SCR反應床的布置。脫氮反應器的安裝位置有多種可能。圖8-12是煙氣脫硝SCR系統安裝位置示意圖[74],催化反應器在鍋爐尾部煙道中布置的位置,有三種可能的方案:

圖8-12 煙氣脫硝SCR系統安裝位置示意圖[74]
① 高溫高粉塵布置。該方式布置在空氣預熱器前溫度約為350℃的位置,此時煙氣中所含有的全部飛灰和SO2均通過催化反應器,反應器的工作條件是在“骯臟”的高塵煙氣中。由于這種布置方案的煙氣溫度(300~400)℃,適合于多數催化劑的反應溫度,因而應用最為廣泛;② 低粉塵布置。該方式SCR反應器布置在靜電除塵器和空氣預熱器之間,溫度為(300~400)℃的煙氣先經過電除塵器后再進入催化反應器,可以防止煙氣中的飛灰對催化劑的污染和將反應器磨損或堵塞,但煙氣中的SO2始終存在,因此煙氣中的NH3和SO3反應生成硫酸銨而發生堵塞的可能性仍然存在。采用這一方案的最大問題是,常規靜電除塵器無法在(300~400)℃正常運行,需要高溫靜電除塵器,因此很少被采用;③ 尾端布置。該方式SCR反應器布置在除塵器和濕法煙氣脫硫裝置(FGD)后,催化劑完全工作在無塵、無SO2的“干凈”煙氣中。由于不存在飛灰對反應器的堵塞及腐蝕問題,也不存在催化劑的污染和中毒問題。這一布置方式的主要問題是,當將反應器布置在濕式FGD脫硫裝置后時,其排煙溫度僅為(50~60)℃,因此,為使煙氣在進入催化劑反應器之前達到所需的反應溫度,需要在煙道內加裝燃油或燃燒天然氣的燃燒器,或蒸汽加熱的換熱器以加熱煙氣,從而增加了能源消耗和運行費用。
(2) 制氨系統。在SCR系統中,靠氨與NOx反應達到脫硝的目的。穩定、可靠的氨系統在整個SCR系統中不可或缺。制氨一般有尿素、純氨和氨水等[75]。
(3) 影響SCR脫硝率的主要因素。催化劑是SCR系統中最關鍵的部分,理想條件下催化劑的壽命可以無限長,但實際上許多因素都可以導致催化劑活性降低。催化劑類型、結構和表面積都對脫除NOx效果有很大影響。此外,在SCR系統設計中,最重要的運行參數是煙氣溫度、煙氣流速、O2濃度、水蒸汽和SO2的存在、鈍化影響和氨滑移等。
(4) 煙氣脫硝SCR技術在國內外的應用和實例。1975年日本在Shimoneski電廠建立了第一個SCR系統的示范工程[76],其后SCR技術在日本得到廣泛應用。SCR技術在日本的運行結果顯示了良好的性能和較高的脫硝率,引起歐洲各國的極大關注,并在歐洲國家迅速推廣。
8.3.2煙氣催化脫硫
按照催化氧化還原機理,可以將催化脫硫分為兩種途徑:一種途徑是利用催化劑將SO2氧化為SO3,SO3可以制硫酸,該途徑稱為催化氧化法;另一種途徑是利用催化劑將SO2還原為單質硫,這種方法可副產硫磺,稱為催化還原法。
8.3.2.1SO2的催化氧化
SO2分子和O2分子直接反應的速率很慢,均相氣態反應的活化能很高,甚至在800℃的高溫也難以進行,因此,SO2氧化反應必須在有催化劑的條件下才能進行。在催化劑作用下,煙氣中的SO2同O2反應生成SO3,然后再把SO3用H2O吸收轉化為稀硫酸或與其他化合物反應轉化為所需的產品。下面按催化劑類型介紹催化氧化工藝:
(1) 釩系催化劑。SO2氧化用的催化劑大都是以釩的氧化物V2O5為催化劑的活性組分,以堿金屬硫酸鹽如K2SO4、Na2SO4或焦硫酸鹽為助催化劑,以硅藻土(或加少量的鋁、鈣、鎂等)為載體,通常稱為釩-鉀-(鈉)-硅體系催化劑。釩系催化劑是目前工業應用比較成熟的催化劑,國外釩催化劑制造企業生產的催化劑載體均采用美國賽力特公司的硅藻土,該公司按產地、硅藻種屬以及硅藻土孔容、孔徑、生產工藝將硅藻土分成不同的牌號出售。國外很多公司都有自己的專利產品,例如丹麥的托普索公司開發生產的VK系列,美國孟山都環境化學公司的Cs系列,德國巴斯夫公司的含銫釩催化劑,德國魯奇公司開發的在二氧化硅或沸石載體上負載氧化鐵和釩的新型催化劑。
(2) 銅系催化劑。銅系催化劑是由氧化銅負載在載體上構成,根據載體的不同,銅系催化劑主要有CuO/AC和CuO/γ-Al2O3兩種。可再生鋁基氧化銅干法煙氣脫硫的原理為:煙氣流過反應器(位于低溫省煤器后)內的氧化鋁載體顆粒,煙氣中的SO2與負載在氧化鋁上的氧化銅發生反應生成CuSO4[(300~500)℃],從而達到脫除煙氣中SO2的目的。
(3) 活性炭。活性炭煙氣脫硫方法具有脫硫效率高、工藝連續的特點,但由于吸附材料價格較高,限制其推廣應用。近年來,利用活性炭纖維、沸石、樹脂、氧化鋁等材料作為吸收劑以及變壓吸附等均有突破性進展。由于活性炭的內表面積較大(活性炭的外表面積與內表面積相比非常小),因此催化反應主要發生在內表面的活性中心。活性炭吸附脫硫是多步復雜的過程,包括SO2、水蒸汽和O2在活性炭表面的吸附、SO2催化氧化生成SO3并進一步生成H2SO4等。脫硫效果的好壞主要取決于活性炭的催化活性,只有具有較高催化活性的活性炭才能達到理想脫硫效果。在活性炭催化活性一定的前提下,水蒸汽、O2的體積分數、反應溫度等對脫硫效果都有較大影響。
(4) Mg/Al/Fe復合氧化物催化劑。Mg/Al/Fe復合氧化物脫硫是一種氧化和吸附的耦合機理,具體過程為:先將SO2氧化成SO3再吸附生成硫酸鹽,吸附飽和后的Mg/Al/Fe復合氧化物可以用H2、CH4或CO還原硫酸鹽再生,高濃度的再生產物SO2和H2S可回收利用。
(5) 液相催化氧化催化劑。SO2的液相催化氧化包括化學吸收和催化氧化兩大過程,化學吸收是固硫過程,催化氧化則是脫硫過程。液相催化氧化法是在水溶液中加入氧化催化劑,使SO2在液相中被催化氧化制取稀硫酸、石膏、N-P復合肥料和聚合硫酸鐵等多種副產品。該法避免了復雜的吸附、脫附步驟,回收工藝簡單。
8.3.2.2SO2的催化還原
催化還原法是SO2在還原劑的作用下直接還原成固態硫,比SO2催化氧化成SO3再吸收制取稀硫酸的工藝簡單,而且副產品硫磺具有易運輸、無二次污染、經濟效益高等多種優越性,因此學術界從20世紀30-40年代就開始探索SO2的催化還原,目前已有許多成功的實驗室催化脫硫方法,但尚未工業化,主要是未克服煙氣中過量氧對還原過程的干擾和催化劑的中毒問題。根據所用還原劑不同,催化還原脫硫可分為H2、CO、CH4、C、合成氣等還原法。
(1) H2還原法。H2作為還原劑,在沒有催化劑的情況下,還原SO2需要在500℃以上才會發生化學反應,而采用催化還原法可使反應溫度大大降低。鋁礬土、Ru/Al2O3、Co-Mo/Al2O3及Fe族金屬負載到Al2O3上的催化劑具有較好的催化還原活性。
H2還原法的優點是操作溫度較低(<300℃),副產物只有H2S,如果通過循環操作,則可使硫的收率進一步提高。缺點是H2的來源、運輸和貯存都不方便,而且煙道氣中含有過量的O2,對反應有較大的抑制作用。此外,H2易爆、易燃,操作危險,脫硫成本偏高,因而難以實現工業化。
(2) CO還原法。CO還原SO2的研究比較深入,研究者已經研制開發出幾十種催化劑,分為負載型金屬氧化物催化劑、鈣鈦礦型復合氧化物催化劑、螢石型復合氧化物催化劑和其他復合氧化物催化劑,并針對不同類型的催化劑提出各種類型的還原脫硫反應機理。用CO還原SO2到單質S所涉及的反應如下:
(8-5)
(8-6)
(8-7)
其中x=2~8或更大。高溫下反應容易發生,生成比SO2更具毒性的COS,因此在反應過程中要盡量減少COS的生成。
① 負載型金屬催化劑。負載型金屬氧化物催化劑一般采用Cu、Fe、Co、Mo、Ni和Cr等過渡金屬負載在Al2O3上制得。Haas L A等[77]研究了Fe/Al2O3催化劑上的反應,認為Al2O3不僅起載體作用,而且和Fe存在協同效應,是雙功能催化劑,與Fe/SiO2比較,發現Al2O3可以催化反應(8-5)的進行。為了減少COS的量,以Fe/Al2O3作第一床層,Al2O3作第二床層,在410℃時SO2轉化率大于90%,COS濃度可降至0.05%;② 鈣鈦礦型催化劑。鈣鈦礦型催化劑用于催化還原SO2一直受到關注,Happel J等[78]最早研究了用鈣鈦礦型催化劑還原SO2,結果顯示,用LaTiO3作催化劑時,COS的生成與SO2轉化率無關,而與CO分壓有關,只要控制CO與SO2比為1.9,就可使COS減小到最少。500℃時SO2轉化率達到95%,而COS僅為0.3%。鈣鈦礦結構作為CO還原SO2的催化劑性能優異,首先在于其抑制COS的生成。但反應后鈣鈦礦結構消失,實際起作用的是金屬硫化物和硫氧化物。鈣鈦礦結構促進了活性相La2O2S和CoS2的生成;③ 螢石型復合氧化物催化劑。螢石型復合氧化物催化劑用于催化還原SO2已經有許多研究進展,Tschope A等[79]研究發現,Cu/CeO2催化劑和復合氧化物Cu-Ce-O都對催化脫硫反應有很高的活性和選擇性。反應溫度大于450℃和CO/SO2為2時,S產率大于95%。
(3) CH4還原法。CH4是天然氣的主要成分,作為還原劑的優點是價廉易得,因此CH4催化還原SO2一直是研究熱點。SO2和CH4之間的基本反應為:
(8-8)
其中[S]代表氣相中不同的硫物種,可以是S2、S6或S8。

8.3.3同時催化脫硫脫硝技術
干法同時脫硫脫硝技術按照氧化和還原反應過程,可分為催化氧化SO2同時還原NOx、同步氧化SO2和NOx以及同步還原SO2和NOx技術。
8.3.3.1催化氧化二氧化硫同時還原氮氧化物
(1) 活性炭加氨法。活性炭用于發電廠煙道氣脫硫脫硝的處理過程分為兩個階段:靜電除塵以后,氣體溫度降至(120~150)℃,利用焦炭的吸附性能吸附SO2,然后以焦炭為催化劑,氨為還原劑催化還原NOx。由于催化反應溫度較低,除塵后的氣體可不必加熱直接處理,節約了能源;同時,活性炭具有范圍極寬的孔徑分布,NH4HSO4等顆粒的沉積問題也不嚴重;此外,在已裝配了濕法脫SO2裝置的系統中,只需附加一個催化還原反應器,即可處理NOx,而不需進行大的設備改造。
(2) CuO氧化還原法。可再生金屬氧化物法脫硫脫硝技術是目前較新的一種脫硫脫硝一體化煙氣凈化技術,應用較多的金屬氧化物為CuO[80]。CuO法吸收還原過程一般采用負載型的CuO作吸收劑,其中以CuO/Al2O3和CuO/SiO2為主,CuO含量通常占4%~6%,在溫度(300~450)℃條件下,與煙氣中SO2發生反應。
(3) 復合金屬氧化物吸附催化法。20世紀90年代,Yoo J S等[81]將Ce加入到尖晶石結構的復合氧化物MgO·MgAl2-xMxO4(M:Fe,V,Cr;x≤0.4),研究了其催化SO2氧化和NOx還原的性能,反應過程如下:
(8-9)
(8-10)
(8-11)
(8-12)
8.3.3.2同時催化氧化二氧化硫和氮氧化物
(1) Pt/BaO/Al2O3吸附氧化。使用Pt/BaO/Al2O3吸附還原技術可以進行同時脫硫脫硝,其機理是:在催化劑上,Pt提供了NOx和SO2的氧化活性位,BaO主要作用是儲存NOx和SO2;NO和SO2在貴金屬Pt活性位上氧化后生成的NO2和SO3從貴金屬上遷移到與貴金屬鄰近的儲存組分BaO上,并與BaO反應生成硝酸鹽和硫酸鹽。由于硫酸鹽比硝酸鹽穩定,SO2占據了NOx的儲存點后會降低NO2的儲存能力,因此共存的SO2將大大降低催化劑對NO2儲存能力。在BaO吸附位上,NO/SO2要大于5時才能有效地吸附NOx。吸附飽和后的Pt/BaO/Al2O3材料先升溫脫除吸附的NOx,然后通過還原性氣體把吸附的硫酸鹽還原后去除。
(2) Na2CO3/Al2O3吸附氧化NOXSO工藝采用負載在高表面積氧化鋁小球上的Na2CO3吸收劑同時吸收SO2和NOx,NOXSO過程的脫硫脫硝率可分別達到97%和70%。
NOXSO工藝的優點是能同時高效去除SO2和NOx,并副產有用的硫磺或硫酸。與傳統的脫硝(如SCR技術)和脫硫技術相比,除凈化效率更高以外,還是一種干式的可再生過程,沒有淤泥和廢液的排放問題;規模可大可小,適應性強,不受電廠操作條件變化的影響,還可用于老廠的改造。
8.3.3.3同時催化還原氮氧化物和二氧化硫
同時催化還原方法是最理想的干法脫硫脫硝技術,采用還原性氣體將NOx和SO2選擇性催化還原為氮氣和單質硫,可避免目前脫硝脫硫工藝冗長的問題,既消除了煙氣中的NOx和SO2,又回收了產品固態元素硫。目前該方法待解決的主要問題有:一是優化還原劑H2、CO、C、CH4、NH3等與催化劑的匹配技術,二是煙氣中的過量氧對還原過程的干擾問題和催化劑的中毒問題。目前的研究只有以CO作還原劑的同時催化還原法。
除上述介紹的各種脫硫脫硝技術外,光催化氧化還原法是近10年來發展起來的一種節能型高效凈化污染物處理工藝,常用的催化劑有TiO2和CdS等。TiO2是性能良好的半導體催化劑,在波長相當于或小于380 nm時,能被激發活化,起催化降解作用。光氧化法的原理是基于在光的照射下,光敏半導體上的價帶電子發生帶間躍遷,激發出光電子和空穴,它們可以與吸附于表面的氧、硫等發生作用,從而發生一系列的氧化-還原反應。在半導體催化劑作用下產生的活性自由基能使SO2和NOx分解。
8.4室內空氣催化凈化
城市中人的生活時間約有90%在室內度過,因此室內空氣污染與人的身體健康密切相關[82-83]。室內空氣污染包括物理性污染、化學性污染和生物性污染。隨著國家環境法規的日益嚴格和公眾環保意識的提高,室內空氣污染引發的一系列問題受到越來越多的關注。科研人員開始深入探討室內空氣污染物的來源、危害、對人類健康的影響以及可行的解決途徑。
根據Klepeis N E等的研究[82],可以將室內常見的空氣污染物及其主要來源總結如表8-3所示。從表8-3可以看出,化學性污染和生物性污染最為突出,且人為污染是主要污染源。因此,采取切實有效的措施以控制此兩類污染顯得尤為重要。

表8-3 主要室內污染物[82]
控制室內空氣污染主要有消除污染源、加強室內空氣流通和凈化污染物三種途徑。消除污染源實際操作較為困難。室內通風換氣簡單、經濟,然而現代化的生活方式使室內通風量受到限制,而且在外界大氣污染比較嚴重的地區,采用通風換氣對降低和消除室內污染不再有任何積極作用。因此,通過凈化技術控制室內污染成為改善室內環境的有效手段[83]。
室內空氣凈化技術主要包括物理吸附技術和催化技術[83]。物理吸附技術利用活性炭、硅膠和分子篩等高比表面材料吸附空氣中的污染物,選擇性好,對低濃度污染物清除效率高,且操作方便。缺點是吸附劑需要定期更換,常伴有二次污染。催化技術則一定程度彌補了其缺點。本節將主要介紹光催化技術、熱催化氧化以及低溫等離子體催化凈化技術,同時介紹微生物的常溫催化凈化技術。
8.4.1室內空氣光催化凈化
8.4.1.1光催化原理
光催化是基于光催化劑在光照條件下促進反應進行的催化氧化還原反應。1972年Fujishima A和Honda K發現在受紫外光照射的TiO2-Pt電極對上可以持續發生水的氧化還原反應生成O2和H2[84]。進入20世紀80年代,光催化在環境凈化和有機合成反應中的應用發展迅速,已成為日益受到重視的一項污染治理新技術。
光催化反應機理如圖8-13所示,半導體受到能量大于其禁帶寬度的光輻照時,半導體價帶(VB)中的電子會吸收光子的能量,躍遷到導帶(CB),從而在導帶產生自由電子(e-),同時在價帶產生空穴(h+),該過程為價帶電子的光激發過程。而激發的電子和空穴可分別參與還原反應和氧化反應。

圖8-13 光催化空氣凈化作用機理示意圖[85]①光激發電子躍遷;②電子和空穴的復合;③價帶空穴氧化吸附物的過程;④導帶電子還原表面吸附物;⑤進一步的熱反應或光催化反應;⑥半導體表面懸掛空鍵對導帶電子的捕獲;⑦半導體表面鈦羥基對價帶空穴的捕獲
根據激發過程,禁帶寬度直接決定了光催化劑能夠吸收利用光的最長波長。禁帶寬度足夠低時,光催化劑才可能有效利用可見光成分。Yu C等[86]提出,合適的光催化劑必須具有如下條件:具有光催化活性(即價帶和導帶位置與反應體系匹配);最好能吸收可見光或至少吸收紫外線(禁帶寬度適合);呈現光蝕惰性及生物惰性;最好廉價。
8.4.1.2常見光催化劑
光催化劑多為半導體,研究最為廣泛的光催化劑為TiO2,其他一些常見的光催化劑還包括SrTiO3、GaAs、MoSe2、CdS、WO3等[87],均為典型的半導體材料。近年來,含Fe的鐵氧體材料也受到較大關注[88],包括BaFe2O4、CoFe2O4、NiFe2O4、ZnFe2O4、CaFe2O4、MnFe2O4、CuFe2O4、Fe3O4等。幾種鐵氧體與其他常見光催化劑價帶、導帶位置相當,決定了其潛在的應用前景。鐵氧體相比于TiO2,禁帶寬度更窄,因而能有效利用豐富的可見光資源。此外,鐵氧體一般具有良好鐵磁性,對于其固定、脫離污染體系等均更易操作。
8.4.1.3光催化凈化室內污染物
光催化劑廣泛應用于室內空氣凈化方面,由于實驗條件溫和,而具有良好的應用前景。
Sano T等[89]對不同貴金屬(Pt,Pd,Ag)負載的TiO2(P25)光催化凈化乙醛進行了研究。如圖8-14所示,濕度在50%時,Pt的添加大大提高了純TiO2的光催化活性,這是由于水分子的存在促進了O2-、OH自由基的形成。Sinha A K等[90]發現,在室溫條件下,Pt/CeO2-TiO2具有較好光催化去除甲苯的活性。

圖8-14 貴金屬(Pt,Pd,Ag)負載的TiO2(P25)光催化凈化乙醛性能[89]
8.4.2室內空氣常溫催化凈化
從原理上講,現有的VOCs催化燃燒技術與室內VOCs的凈化沒有本質區別。其關鍵差異在于室內空氣凈化需要室溫常壓環境,對催化劑性能提出更高要求。目前,已成功研制出可室溫條件下催化凈化CO、甲醛的催化材料,并在室內空氣凈化方面展現出良好的應用前景。
8.4.2.1常溫催化凈化室內一氧化碳
目前,可室溫、甚至0℃以下催化氧化CO的催化劑主要有兩類,分別是以Au為代表的貴金屬催化劑和以Co3O4為主的金屬氧化物催化劑。
(1) 金催化劑。Au一直以來都被認為是沒有催化活性的惰性金屬,但20世紀80年代年后期,Haruta M等[91]發現負載在過渡金屬氧化物上的納米Au催化劑對CO低溫氧化具有很高的催化活性。目前,關于金屬氧化物負載的(2~4) nmAu催化劑具有高催化氧化CO活性的論斷已基本達成共識,雖然納米效應的起源目前還存在一定爭議。另一方面,具有活性的Au的價態也存在一定爭議。
(2) 金屬氧化物催化劑。貴金屬催化劑廣泛應用于CO催化氧化,由于價格高昂限制了其廣泛應用,研究者一直在尋求開發能夠替代貴金屬的非貴金屬催化劑,其中Co3O4以其優異的低溫催化CO氧化性能成為最具應用前景的非貴金屬催化劑。
8.4.2.2常溫催化凈化室內甲醛和VOCs
初期研究主要集中于甲醛催化氧化上。Sekine Y等[92]對Ag2O、PdO、Fe2O3、ZnO、CeO2、CuO、MnO2、Mn3O4、CoO、TiO2、WO3、La2O3和V2O5等金屬氧化物室溫下對密閉體系中甲醛的分解進行了研究(見表8-4),發現MnO2室溫下可氧化分解甲醛為CO2和H2O,有望作為凈化室內甲醛材料的活性組分。

表8-4 室溫下金屬氧化物對甲醛的分解活性對比[92]
①為粗制樣品
貴金屬催化劑是目前最接近室溫條件催化氧化甲醛的催化劑。最近,Zhang C B等[93]在高效室溫氧化甲醛催化劑研究方面已經取得突破,開發出可室溫催化氧化甲醛的Pt/TiO2催化劑。
到目前為止,利用催化氧化技術僅僅實現了對甲醛的室溫催化氧化,而針對室內其他主要VOCs如乙醛、環己酮以及苯系物等的催化氧化在室溫下還難以實現,在眾多應用于醛酮類和苯系物催化氧化的貴金屬、過渡金屬氧化物催化劑中,完全分解上述污染物的最低反應溫度分別在200℃和150℃以上。另外,從研究現狀和發展趨勢看,開發可室溫催化氧化室內其他有機污染物的催化材料也具有很大難度。
8.4.3低溫等離子體協同催化技術
近年來興起的低溫等離子體催化技術(non-thermal plasma catalysis)是一種新興的技術,結合了低溫等離子體和催化反應的優點,在有效彌補兩種凈化技術不足的同時,充分發揮了催化劑和低溫等離子體之間的協同作用[94],在環境污染物處理方面引起極大關注,被認為是環境污染物處理領域很有發展前途的高新技術之一[95],有望實現在室內VOCs凈化中的實際應用。
8.4.3.1低溫等離子體產生方式
低溫等離子體主要是通過氣體放電產生,目前利用的主要是介質阻擋放電(Dielectric barrier discharge)。介質阻擋放電產生于由電介質隔開的兩個電極之間,當兩極間加上足夠高的交流電壓時,電極間隙的氣體會被擊穿而產生放電。介質阻擋放電結合了輝光放電和電暈放電的優點,具有電子密度高和可在常壓產生大面積的低溫等離子體的特點,具有大規模工業應用的可能性。
8.4.3.2低溫等離子體協同催化作用機理
將催化劑引入低溫等離子體,則低溫等離子體和催化反應之間存在協同作用。一方面,在低溫等離子體空間內富集了大量極活潑的如離子、電子、激發態的原子、分子及自由基等含有巨大能量的高活性物種。活性粒子一方面活化了反應分子,另一方面活化了催化劑中心。因此,可使常規條件下需要很高活化能(加熱到300℃以上)才能實現的催化反應在室溫條件下即可順利進行,大大減少了能耗。另外,催化劑的存在還可促進等離子體產生的副產物完全氧化和臭氧分解反應,消除二次污染[95-96]。必須指出,低溫等離子體和催化劑之間的相互作用十分復雜,關于二者協同作用的機理并沒有非常明確的解釋,還需要更加深入的研究。
8.4.3.3低溫等離子體催化凈化室內VOCs
多種催化劑已用于低溫等離子體催化反應,主要包括光催化劑、金屬氧化物催化劑、貴金屬催化劑及分子篩類等。典型的催化劑有TiO2、MnO2、Pt/Al2O3、Al2O3、鐵錳氧化物、ZSM-5和CoOx等。
Subrahmanyam C H等[97]設計了一種新式介質阻擋低溫等離子體催化反應器,反應器結構如圖8-15所示,采用其研究甲苯凈化,甲苯轉化率如圖8-16所示。可以看出,在235 J·I-1的能量輸入密度下,對于負載有Mn或Co氧化物的SMF,甲苯轉化率可以達到100%。

圖8-15 新式介質阻擋低溫等離子體催化反應器[97]

圖8-16 低溫等離子體催化凈化甲苯性能[97]
在放電等離子體處理VOCs的過程中,臭氧作為強活性氧化物質對VOCs的氧化降解起積極作用,但若降解后最終排放氣的臭氧濃度過高,也將造成空氣污染。Futamura S等[96]研究發現,MnO2能夠加速O3向O2的轉化,可以作為放電等離子體反應器的后處理改善最終的排氣品質,并且轉化過程生成的活性氧物種可能對MnO2分解VOCs起到作用。
總之,該技術本身還存在一些急需解決的問題。首先,該復合技術的理論還不完全成熟,其次,設備制造技術難度大、成本費用高、價格貴。另外,盡管低溫等離子體基本上安全,但實際應用還存在一定的安全隱患,如輻射等。
8.4.3.4常溫催化凈化室內微生物
常溫催化凈化室內微生物可以根據是否利用光照條件分為光催化技術和非光催化技術。
(1) 光催化凈化室內微生物污染。構成微生物的有機物化學鍵主要為O—H、C—H、N—H以及O—P鍵等。從理論上講,只要光催化產生的自由基氧化能力大于這些化學鍵的鍵能,就可以達到殺菌的目的,這與光催化氧化VOCs的機理相同。光催化體系利用各種途徑的紫外光產生的·OH具有極強的氧化能力,其氧化作用幾乎無選擇性,且能夠穿透細胞膜破壞細胞膜結構,阻止成膜物質的傳輸,阻斷其呼吸系統和電子傳輸系統,在室溫條件下即可將室內空氣中的病毒、細菌等微生物滅活,甚至導致細胞完全礦化[98]。
(2) 非光致催化凈化室內微生物污染。光催化雖然是一種比較高效的室內空氣常溫消毒手段,但該技術對太陽光利用率低,應用較難。實際上現在日常使用最為廣泛的還是化學消毒劑,開發無毒無害、長效安全的新型抗菌材料和技術是十分迫切的任務,也是當前和今后抗菌領域的重要研究課題和發展方向[99]。

8.5水處理過程中的多相催化
隨著化學工業的高速發展,出現了很多難降解的有毒有機污染物,如農藥、醫藥品類化合物、內分泌干擾物、染料等。現有的廢水處理工藝如吸附、絮凝、臭氧氧化和生物氧化技術均不能有效去除這類污染物,它們被直接排放或通過地表徑流進入淡水體系,致使水環境都遭受不同程度的污染。我國水資源不足、分布不均勻、利用率很低。據統計,我國669個城市中有400個城市供水不足,110個城市嚴重缺水。水環境污染出現惡化趨勢,水質富營養化、有機物、氨氮以及病源性微生物嚴重超標,人類的身體健康受到極大的威脅。面對國家在飲用水安全保障以及水質改善方面的重大需求,亟待研究經濟高效高新的水處理技術,解決水質污染問題。
目前,水質催化凈化技術有可能處理所有類型的有機和無機污染物,這些催化凈化技術包括氧化和還原過程。氧化過程是基于氧化劑的過程,通常稱為高級氧化過程,主要包括光催化氧化、芬頓催化氧化、臭氧催化氧化和濕式催化氧化。這些氧化過程主要以光、H2O2、O3等物理、化學氧化劑為媒介,協助多相催化氧化,產生羥基自由基、超氧自由基等強氧化活性物種,氧化分解結構穩定的有機物,獲得無機礦化或者提高有機物的可生化性,與生物氧化處理工藝結合,對高濃度有機廢水達到凈化目的。還原過程是利用金屬催化還原水體中的無機污染物,達到脫毒的目的。針對地下水中硝酸鹽的去除,可以通過雙金屬催化還原的方法,將硝態氮轉化為無毒的氮氣。本節立足于水中難降解有機污染物和硝酸鹽的去除,介紹光催化、芬頓催化氧化、臭氧催化氧化、濕式催化氧化以及雙金屬催化還原等水處理技術原理與應用,重點闡述催化氧化和催化還原降解污染物的過程以及催化材料的研究進展,進一步提出每種催化技術存在的優勢與不足以及未來發展的方向。
8.5.1光催化水處理技術
光催化是一種具有成本效益的高級氧化技術,特別是對持久性有毒有機污染物的催化降解,并且有望用太陽能作為反應光源。光催化過程中,半導體催化劑吸收高于其禁帶寬度的光能后,價帶中的電子受激發,形成電子/空穴對,在電場作用下,電子與空穴發生分離,并與吸附在催化劑表面的O2或者H2O發生反應形成活性氧自由基,這些活性氧自由基能氧化大多數的有機污染物及部分無機污染物。TiO2、ZnO、SrTiO3、CeO2、WO3、Fe2O3和ZnS等因其滿價帶空導帶的特殊電子結構而作為光催化劑用于氧化還原和電子轉移過程中。其中TiO2因其化學穩定性高、耐腐蝕,且具有較深的價帶能級,催化活性好,無毒無害和成本較低而成為最為廣泛應用的光催化材料。然而,對于TiO2光催化體系中的大多數降解反應,量子產率低于10%。通過陰陽離子摻雜可以提高TiO2的光吸收和光催化活性。在銳鈦礦相TiO2表面摻入貴金屬如Pt、Pd、Ir和Ag等能夠顯著提高其光催化活性,這些貴金屬是電子捕獲中心。銳鈦礦相氧化物也可以與WO3、SnO、ZrO2或其他體系建立異質結,促進電子空穴分離過程。在自然界中,輻射到地面的紫外光能量約占太陽光總輻射能量的5%。基于TiO2的催化劑對可見光的響應效率低,為了擴大光譜利用范圍,充分利用全波譜太陽能,多種多樣的新型可見光催化劑涌現,主要有鈣鈦礦(A2+B4+O3)及其相關結構材料、白鎢礦(A3+B5+O4)和尖晶石型材料(AB2O4)。特別是類鈣鈦礦型化合物具有穩定的結構,可以與一系列金屬離子形成固溶體,成為可取代TiO2的具有應用前景的可見光催化劑。鈣鈦礦結構的材料主要有鉍酸鹽化合物MBiO3(M=Li、Na、K、Ag)和鐵酸鹽化合物(如BiFeO3)。這些材料一般通過水熱法合成,通過控制水熱合成的參數可以得到不同的形貌。Ruan Q J等[104]指出,不同的形貌發生不同的光催化反應,微米盤和納米片在紫外輻射下都具有光反應活性,而納米片在可見光輻射下具有更高的光催化活性。鈣鈦礦相關結構材料中,Bi2WO6是最簡單、研究最為廣泛的催化材料,Bi2MoO6和PbBiO2Cl也具有可見光催化活性。與之相關的鹵氧化物在可見光范圍也具有光活性,溴氧化物和碘氧化物,如BiOBr、PbBiO2Br、BiOIxBr1-x或BiOIxCl1-x都具有良好的可見光響應能力。另外,有研究表明,納米金或銀與半導體復合可以形成具有Plasmon效應的可見光催化劑。Hu C等[105]以介孔氧化鋁為載體,成功研制出新型高效的Ag-AgI/Al2O3可見光催化劑,將納米銀的Plasmon共振效應引起很強的可見光吸收應用到光催化降解中,并且發現納米銀與鹵化銀半導體匹配,受光激發引發兩個界面電子轉移過程:一個是納米銀到鹵化銀的導帶,另一個是從水中污染物到表面光致納米銀離子,產生了超氧自由基和光致納米銀空穴兩種活性物種,二者的協同產生強氧化能力,并保證了納米銀與鹵化銀的光穩定性(見圖8-17)。
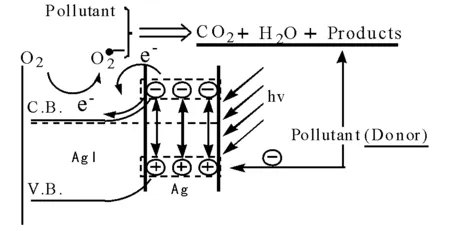
圖8-17 Ag-AgI/Al2O3的光催化機理示意圖[105]
在光催化降解污染物的過程中,O2參與了污染物轉化及礦化的整個反應過程,如羥基化、開環和脫羧等,污染物降解反應的最終凈結果為污染物與O2反應生成H2O和CO2,期間伴隨著各種含氧活性物種的生成及反應,因此,光催化活化O2的微觀機理是該領域的一個重要基礎科學問題。研究者利用同位素標記等手段研究了TiO2光催化活化O2的微觀過程,發現反應物分子中的氧原子在轉化過程中完全被O2中的1個氧原子所置換(置換率>99%),提出了與以往貴金屬等催化氧化機理完全不同的TiO2光催化氧原子轉移機理[105-106]。進一步的研究闡明了芳環類污染物光催化羥基化過程中O2與H2O的活化機理與參與途徑[107]。另外,作為一種多相催化反應,光催化劑結構對催化性能存在明顯影響。研究者發現不同晶面結構對光催化降解過程中O2的活化有顯著影響,隨著銳鈦礦相TiO2{001}晶面比例的提高,中間產物中羥基氧來源于O2的比例上升,相同污染物轉化率的條件下,礦化率提高,與此同時轉化單位污染物所消耗的O2量上升,這主要與O2在不同晶面上的吸附能力及光生電子產生速率不同有關[108-109]。
光催化作為一種高級氧化技術被廣泛研究用于太陽能轉化和水質凈化。通過紫外或可見光催化進行水處理,在液固界面產生羥基自由基,隨后的自由基反應服從異相反應動力學。目前光催化的主要缺陷是量子產率低,不能對所有的水質進行有效地凈化。然而,強大的理論支持和應用性促進光催化的發展,太陽光助水環境修復是光催化的主要應用領域,并將繼續作為光催化技術的重要實踐平臺和建立相關理論知識的動機。
8.5.2綠化催化新工藝——芬頓技術的發展及應用
芬頓(Fenton)試劑一般是指Fe2+與H2O2構成的體系,由法國Fenton于1894年在H2O2氧化蘋果酸的實驗中發現[110],Haber-Weiss于1934年提出芬頓反應的羥基自由基機制,指出了Fe2+在反應過程中的催化作用,Fe2+/Fe3+的電子轉移催化分解H2O2產生強氧化性的羥基自由基,如式8-13和式8-14所示。羥基自由基的氧化電位高達2.8 V,而且反應沒有選擇性,能夠將有機污染物分解為小分子有機物或部分礦化為CO2和H2O。Eisenhouser H R于1964年首次將芬頓試劑應用于處理苯酚和烷基苯廢水[111]。芬頓氧化符合目前國際社會所倡導的綠色化學理念,能夠在環境友好的氧化劑——H2O2存在條件下,將有毒或難降解的有機污染物礦化為對環境無污染的CO2和H2O,是一種環境友好的綠色催化新工藝。
k1=63 mol-1·s-1
(8-13)
k2=0.02 mol-1·s-1
(8-14)
均相芬頓反應具有高效、成本低廉的優點,但其反應效率受溶液中亞鐵離子濃度和亞鐵離子再生能力的影響,而且在反應過程中產生大量含鐵污泥,處理成本較高,很容易造成二次污染,成為制約其實際應用的重要因素[112]。非均相芬頓催化能夠克服這些缺陷,這是因為非均相芬頓催化拓寬了pH適用范圍,而且反應后催化劑可以回收再利用,降低了廢水處理成本,具有廣泛的應用前景。非均相芬頓催化劑主要分為過渡金屬或過渡金屬氧化物復合體、負載型芬頓催化劑。
過渡金屬,如Fe0在酸性條件下可以和H2O2反應產生Fe2+并促進Fe2+再生。國外有研究者認為Fe0/H2O2體系無論是處理效果,還是運營成本均優于Fe2+/H2O2體系[113-114]。過渡金屬氧化物也可以催化H2O2產生羥基自由基,從而降解水體中的有機污染物。以典型的鐵氧化物為例,如磁鐵礦、赤鐵礦、針鐵礦和水鐵礦等均可直接作為非均相芬頓催化劑催化降解偶氮染料、喹啉和單萜烯等。為了提高催化劑的催化活性,許多研究通過摻雜其他的過渡金屬陽離子或者H2熱處理還原等方式對鐵氧化物結構進行修飾[115]。Nie Y等[116-117]研究了Cu摻雜α-FeOOH、Bi2O3和LaTiO3鈣鈦礦的催化活性及催化機理,研究表明,這些催化劑均能催化H2O2產生羥基自由基,活性金屬不同價態間發生電子轉換,可以循環高效地去除水體中染料、內分泌干擾物、醫藥品等有機污染物。
負載型催化劑是另一種具有應用前景的非均相芬頓催化劑,活性組分包括過渡金屬離子、過渡金屬氧化物以及以過渡金屬離子為中心的金屬有機化合物;載體的選擇有多種類型,包括無機和有機載體,如碳材料、無機氧化物、黏土礦物、陽離子交換樹脂等。Navalon S等[118]將Au負載于芬頓反應處理的金剛石,此催化劑能有效去除苯酚,在pH=4的條件下,催化效率比其他固體催化劑高4個數量級。Xu L等[119]最近采用浸漬法成功地研制出磁性納米型Fe3O4/CeO2復合催化劑,并研究了該催化劑對4-氯酚的多相芬頓催化降解效果,結果顯示,在pH=3.0和溫度30℃條件下,Fe3O4/CeO2(2.0 g·L-1)和H2O2(30 mmol·L-1)對4-CP(0.78 mmol·L-1)降解的擬一級動力學常數為0.11 min-1,該催化劑對H2O2的利用率高達79.2%。
芬頓催化氧化技術具有操作過程簡單,無需復雜設備且對環境友好等優點,已逐漸應用于染料、農藥等廢水處理工藝中,具有很好的應用前景和極大的推廣價值。但從現有國內外的研究成果看,芬頓催化氧化技術尚存在氧化降解能力需要提高、污染物礦化速率偏慢、需要紫外光、超聲等外能輔助、粉末催化劑難以回收等缺點,因此,改善芬頓反應羥基自由基的產生機制和反應條件,提高羥基自由基生成率和利用率,設計結構簡單、效率高、可長期穩定運行的反應器將是該技術發展的必然趨勢。
8.5.3臭氧催化氧化水處理技術
臭氧催化氧化技術是近年來發展起來的一種具有較強競爭力的高級氧化技術,可以在常溫常壓下高效地降解礦化難降解的有機污染物,具有催化效率高、穩定性好、不引入二次污染、催化劑可再生的優點。在酸性介質中,臭氧在室溫時的標準氧化還原電位為2.07 V,是自然界較強的氧化劑之一。臭氧催化氧化技術應用最廣泛、最成功的領域是飲用水的處理。臭氧用于飲用水處理,主要有殺菌消毒、無機物和有機物的氧化、控制氯化消毒副產物和藻類、助凝等功能[120]。非均相催化臭氧氧化的催化劑主要有金屬氧化物和負載型金屬氧化物兩類。
非均相催化臭氧氧化中常用的金屬氧化物催化劑有MnO2、Al2O3、TiO2、CeO2、ZnO和FeOOH等。其中,MnO2因表現出最好的催化臭氧化活性、可有效催化降解的有機物種類最多而受到廣泛關注。MnO2的活性隨pH的降低而提高,催化劑結構、合成方法也是影響活性的主要因素。工業生產的MnO2因粒子較大、比表面積較小幾乎沒有任何催化活性,預合成的以固相形式存在的水合MnO2(水合MnⅣ)比原位形成的MnO2催化活性略低[121]。Lü A等研究發現α-FeOOH、β-FeOOH和γ-FeOOH中,α-FeOOH在催化臭氧氧化降解硝基苯時顯示出最高的催化活性。Co,Mn摻雜α-Fe2O3和Fe3O4也能夠有效地催化臭氧氧化2,4-D、2,4,6-三氯苯酚和安替比林[122-123],多氧化態有助于界面電子轉移,提高催化反應性能,催化臭氧分解形成羥基自由基是加速水中有機污染物去除的主要原因。
金屬氧化物如MnO2、TiO2、Fe2O3、CoOx等固定于硅膠、Al2O3、ZrO2和TiO2等載體上也能作為催化臭氧氧化催化劑催化去除水中有機污染物。這些負載型金屬氧化物的催化活性主要取決于制備方法、受熱歷程、金屬氧化物的性質及其表面特性。Yang L等[124]將MnOx負載于介孔ZrO2或Al2O3、將CoOx負載于ZrO2,研究其在去除水中除草劑2,4-D和醫藥品類如安替比林、布洛芬等過程中的催化活性,結果表明,MnOx和CoOx的多氧化態和高分散性增強其界面電子轉移速率,提高催化活性,羥基自由基的形成導致有機污染物的礦化降解,如圖8-18所示。 介孔Al2O3上負載β-FeOOH導致表面Lewis酸位增強,能使表面產生更多的化學吸附水,增強與臭氧的相互作用,能夠有效地催化臭氧氧化水溶液中布洛芬和環丙沙星[125]。

圖8-18 臭氧催化劑MnOx/介孔Al2O3對不同有機污染物的礦化[124]
堇青石、鈣鈦礦、沸石和蜂窩陶瓷普遍應用于催化臭氧化中催化降解硝基苯、苯甲酮、酚類化合物等。Zhao L等[126]研究發現,蜂窩陶瓷是催化臭氧氧化硝基苯的活性催化劑且反應過程中涉及羥基自由基。進一步對蜂窩陶瓷進行修飾,發現負載Mn、Cu或K后,羥基自由基產生明顯加速。反應機制是中性的表面鍵合羥基促使臭氧分解形成羥基自由基,從而加速硝基苯的催化降解。Beltrán F J等[127]研究指出,LaTi0.15Cu0.85O3鈣鈦礦和活性炭能更有效地催化去除含醫藥品水中的總有機碳(TOC)。鈣鈦礦催化臭氧氧化的控制步驟是化學反應(均相和異相反應);而對于活性炭,外部擴散(臭氧擴散到活性炭表面)是控制步驟。此外,沸石和火山巖在催化臭氧氧化沒食子酸時也顯示出很高的催化活性。
非均相催化臭氧氧化作為有效的深度處理技術,能進一步去除TOC,滿足日益嚴格的出水排放標準,日漸受到關注。該項技術還有以下進步空間:首先,臭氧在水中溶解度較低,提高臭氧利用效率已經成為該技術研究的熱點;其次,研制催化效果好、壽命長、重復利用率高的催化劑,提高臭氧催化分解生成羥基自由基的效率;再次,研究臭氧與其他技術的聯合,降低物耗和能耗,降低污水處理成本也是今后研究工作的重點。
8.5.4濕式催化氧化技術
濕式催化氧化技術(Catalytic Wet Oxidation,CWO)是指在高溫[(200~280)℃]、高壓[(2~8) MPa]下,以富氧氣體或氧氣為氧化劑,利用催化劑的催化作用,加快廢水中有機物與氧化劑間的反應,使廢水中的有機物及含N、S等的毒物氧化成CO2、N2、SO2、H2O,達到凈化之目的。該技術專門用于高濃度工業廢水的處理,是一種廢水深度處理技術[128],表8-5為濕式催化氧化技術處理部分行業高濃度工業廢水的試驗結果。發達國家將濕式催化氧化技術視為第二代工業廢水處理高新技術,專用于解決第一代常規技術(如生物處理、物理化學處理等)難以解決或無法解決的凈化處理問題。濕式催化氧化技術將成為21世紀工業廢水處理的替代新技術。
濕式催化氧化技術的最初研究集中在均相催化劑。均相濕式催化氧化技術是向反應溶液中加入可溶性的催化劑,以分子或離子水平對反應過程進行催化。均相催化的活性和選擇性可以通過配體的選擇、溶劑的變換及促進劑的添加等因素精細的調配和設計。常用過渡金屬(如Co、Cu、Ni、Fe、Mn、V等)的鹽可作為催化劑,但由于均相催化劑溶于廢水出水中,為避免催化劑流失以及對環境的二次污染,需要進行后續處理,同時也提高了廢水處理的成本,使處理工藝的實用性較差,較難實現工業化應用。
從20世紀70年代后期,濕式催化氧化反應的研究重點轉移到非均相濕式催化氧化反應。催化劑在非均相濕式催化氧化反應過程中以固態存在,催化劑和廢水的分離比較簡便,使處理流程大大簡化;此外非均相濕式催化氧化催化劑還具有催化活性高,穩定性好等特點。因此,開發高活性、高穩定的固態催化劑是濕式催化氧化法進行工業化生產的關鍵。非均相催化劑主要有貴金屬系列(如Ru、Rh、Pd、Pt等)和非貴金屬系列(如Cu、Mn、Ce、Mo等),其中貴金屬常以低于5%的負載量負載于γ-Al2O3、TiO2、CeO2、ZrO2和活性炭等載體上;普通金屬常作為活性組分,以氧化物、復合氧化物或負載型催化劑形式應用于濕式催化氧化過程中。例如,Keav S等[129]以鈰氧化物為載體,制備了貴金屬Pt或Ru負載型催化劑,用于濕式催化氧化去除苯酚,在溫度160℃時,苯酚轉化率可達100%。Ayusheev A B等[130]在氮摻雜炭納米纖維上負載Ru為催化劑,進行濕式催化氧化去除苯酚的研究,發現氮摻雜能夠在一定程度提高催化劑活性。貴金屬如Ru、Pt、Pd等在氧化反應中具有高活性和穩定性,但貴金屬價格昂貴,在某種程度上限制其在濕式催化氧化中的應用。常規的金屬如Mn、Ce、Zn、Mo或其氧化物為催化劑的活性已得到證實。例如,Arena F等[131]以MnCeOx為催化劑,研究其在O2壓力0.9 MPa條件下對苯酚、乙酸、草酸、甲酸的降解情況,結果表明,在溫度(110~150)℃,MnCeOx顯示最高的污染物去除率和礦化率。Li Y等[132]在pH=7的條件下,利用浸漬法研制了Mo-Zn-Al-O催化劑,該催化劑在常溫常壓下對陽離子紫XB-L的脫色率達94.6%,TOC去除率可達86.7%。
濕式催化氧化主要有以下優點:① 由于反應在接近絕熱狀態下進行,出口溫度高,停留時間短,氧化反應速率快;② 裝置從靜止到正常運行所需時間很短;③ 工藝過程不受污染水組分改變的影響;④ 占地面積小,可產業化;⑤ 由于反應生成CO2,無二次污染問題;⑥ 可回收熱量;⑦ 處理效率高適應范圍廣;⑧ 處理后生物降解性能提高。目前濕式催化氧化的應用存在較大困難:均相催化劑一般比非均相催化劑活性高,反應速率快,但流失的金屬離子易引起二次污染;在非均相催化劑中,大部分情況下貴金屬的催化活性高,但價格昂貴。普通金屬系列催化劑往往存在溶出問題而限制了其工業化應用,制備高穩定性、高效非均相負載型催化劑,是當今濕式催化氧化研究的熱點和濕式催化氧化工業應用的關鍵。
濕式催化氧化技術今后的發展趨勢為:① 濕式氧化技術是處理難降解廢水的重要方法,應進一步擴大應用范圍,開展濕式氧化與其他處理工藝相結合的廢水處理新工藝,使這一方法在環境治理中發揮更大的作用;② 隨著環保要求的不斷提高,危險廢物處理成了環境研究的熱點和難點,濕式氧化技術將是一個很好的選擇;③ 高效穩定的催化劑以及應用材料方面的研究將是限制催化濕式氧化發展的關鍵因素,因此,制備高效高穩定性的非均相催化劑是濕式催化氧化技術發展的關鍵。
8.5.5雙金屬催化劑催化去除水中硝酸鹽


催化還原是一種很有前景的水處理技術,選擇性地將污染物轉化為無毒或低毒并且易于生物降解的物質。化學催化還原硝酸根指以氫氣、甲酸等為還原劑,在反應中加入適當的催化劑,以減少副產物的生成,也就是利用催化劑的催化作用將硝酸鹽氮還原,反應歷程如下:
(8-15)
(8-16)
目前已經研究開發了幾種催化劑用于催化還原水中的硝酸鹽污染物,如負載Pd、Pt、Cu、Zn等及基于Pd等的雙金屬催化劑。一般來說,基于Pd的催化劑活性更高、更穩定并且選擇性地形成無毒或低毒產物。Pd與其他金屬結合形成雙金屬催化劑,通過改變其幾何形態和電子性質,能夠有效地增強其催化活性。例如,趙偉榮等[135]制備了一系列單金屬或雙金屬催化劑用于水中硝酸鹽的還原,包括負載于Al顆粒上的單金屬Cu、Ni和雙金屬Pd-Cu、Pt-Cu、Au-Cu、Ru-Cu,實驗研究表明,0.4%Pd-4%Cu/Al在pH=4的條件下對硝酸鹽還原形成氮氣具有最高的選擇性(34.1%)和最高的硝酸鹽去除速率(1.41×10-2min-1)。
為了增加金屬分散度和促進相分離,Pd和其他金屬常負載于載體材料上,普遍用于污染物還原的載體有活性炭、氧化鋁和氧化硅。其他不常用但能有效催化還原硝酸鹽和亞硝酸鹽的載體有TiO2、ZrO2、SnO2和有機樹脂、導電聚合物和碳納米管。這些載體通過影響其表面金屬團簇的密度、粒度、形貌和活性位點的分布間接影響催化劑的活性和選擇性,高比表面積或微孔載體因傳質效應而影響其反應活性和選擇性[136-137]。
影響化學催化還原硝酸根的因素很多,包括催化劑的性質(載體、負載量、雙金屬質量比、催化劑制備方法、負載型催化劑用量)、水體因素(反應溫度、pH值)、反應條件(氫氣氣壓或流速)及傳質過程等。其中任一因素發生變化,都會影響硝酸鹽的脫除速率及最終反應物的組成,即影響催化劑活性和選擇性。催化劑活性以單位質量催化劑在單位時間內脫除硝酸鹽氮的量表示;選擇性以某一產物(通常為氮氣)的產率表示。
國內外大多數的硝酸鹽催化加氫脫氮研究都在實驗室進行,實際地下水處理僅限于規模嘗試性研究,大規模運用加氫催化還原脫除硝酸鹽的技術尚不成熟。在實際應用過程中仍可能存在不少問題。因此,在將來的研究中,下列兩個方面應加以重視:
(1) 硝酸鹽還原成N2是一個連續的反應,反應中會生成一些毒性較高的中間產物如亞硝酸鹽、銨根離子和NOx,如何設計反應器使這些中間產物不排放出或控制在排放標準以內,是必須解決的問題。設計兩段反應器,前段用雙金屬負載催化劑僅催化還原硝酸鹽,后段用單金屬負載催化劑來還原亞硝酸鹽,完全或最大限度轉化這些中間產物,可能是一個有效的解決途徑;
8.6溫室效應和臭氧層消耗物質的催化轉化
8.6.1CH4-CO2催化重整
CH4和CO2都是自然界中廉價且資源豐富的含碳化合物,同時也是引起全球氣候變暖的兩種最主要的溫室氣體。隨著全球環保意識的提高以及排放法規的日趨嚴格,如何將CH4和CO2有效消除、處置或資源化利用,引起世界各國的關注。研究者們分別針對CH4和CO2的資源化轉化進行了大量研究,本節將重點對CH4-CO2重整反應進行評述。
8.6.1.1CH4-CO2重整反應的熱力學
CH4-CO2重整反應主要按照(8-17)進行,由熱力學計算可知,CH4-CO2重整反應制取合成氣是強吸熱過程。
△H298=247 kJ·mol-1;
△G0=61 770-67.32T
(8-17)
CH4-CO2重整反應除按照(8-17)反應外,同時存在逆水汽變換(RWGS)(8-18)、CO2歧化反應(8-19)和甲烷裂解反應(8-20)。
△H298=41 kJ·mol-1;
△G0=-8 545+7.84T
(8-18)
△H298=-172 kJ·mol-1;
△G0=-39 810+40.87T
(8-19)
△H298=75 kJ·mol-1;
△G0=21 960-26.45T
(8-20)
除上述四個反應,下面反應也存在于CH4-CO2重整反應中:
(8-21)
8.6.1.2催化劑體系
用于CH4-CO2重整反應的催化劑活性組分主要為Ⅷ族金屬元素,如金屬Ni、Fe、Co、Ru、Rh、Pd、Ir和Pt對催化CH4-CO2重整反應都具有較高活性[138],Os[139]在OsO4/NaIO4水溶液中也能活化甲烷。貴金屬催化劑Pt、Ir、Ru、Rh同時具有較高催化活性和很好抗積炭性能[140]。由于貴金屬資源匱乏,導致其價格高昂,所以非貴金屬催化劑仍是研究熱點。在相同分散度下,Ni基催化劑的轉化率僅低于Pt和Ir,成為最具有可能取代貴金屬的CH4-CO2重整催化劑[141]。
CH4-CO2重整反應除要求高的催化劑活性外,同時由于整個反應在較高溫度下進行,對催化劑的熱穩定性要求很高,必須選擇既具有高溫穩定性同時又具有較高表面積的載體。最初選擇使用的載體多為Al2O3[142]、MgO[143]等,但反應測試表明,Ni負載在該類載體上,催化劑活性迅速降低,催化劑失活的主要原因是Ni顆粒迅速長大和表面積炭。在這個基礎上,增強Ni和載體之間相互作用及具有高耐積炭能力的鎂鋁尖晶石載體成為眾多催化劑選用的載體[144]。不僅鎂鋁尖晶石具有這些優點,同樣能增強Ni和載體之間相互作用及具有高耐積炭能力的載體ZrO2[145]、鈣鈦礦類[146]、分子篩等載體[147]也被發現并報道。
8.6.1.3CH4-CO2重整反應的動力學
在反應過程中,活性金屬組分直接參與了CH4和CO2的活化,因此,其對反應動力學影響顯著。CH4和CO2的活化主要是通過與活性組分的HOMO和LUMO軌道的相互作用進行,活性金屬組分的電子結構是影響反應速率的主要因素。研究發現,CH4和CO2重整反應的轉化頻率(TOF)與活性金屬組分的d特性因子有較好的關聯性[148]。
8.6.1.4反應機理
Wei J M等[149]通過使用同位素和動力學測試,提出Ni基催化劑上CH4-CO2重整反應機理,認為反應過程中沒有通過CHxO含氧物種參與反應這一步,同時認為甲烷活化是反應的速率控制步驟。反應的進程如下:

(8-22)
2[CO2+*?CO2*]
(8-23)
H2+2*?2H*
(8-24)
2[CO2+H*?CO*+OH*]
(8-25)
8.6.2氧化亞氮的催化消除
8.6.2.1氧化亞氮的來源、危害和對策
N2O是一種無色的有微弱甜味的氣體。主要有兩大來源:一是來自自然界,如海洋、森林、土壤等自然源;另一個來源于人類活動。目前對于N2O的催化消除主要有直接催化分解法和選擇性催化還原法。前者是使N2O直接在催化劑上分解為N2和O2,后者主要是通過添加還原劑(如CO、H2、NH3或CH4、C3H6等碳氫化合物)實現對N2O的還原。下面將主要對直接催化分解法方面的研究進展情況進行總結,并在介紹分子篩催化劑體系時對選擇性催化還原法進行簡單回顧,對將來的研究方向和方法提出建議。
8.6.2.2氧化亞氮直接催化分解反應及反應機理
(1) 催化N2O直接分解反應。N2O是一個不對稱分子,N—N的鍵級為2.7,N—O的鍵級為1.6,相比之下N—O鍵更容易斷裂。但N—O鍵能為(250~270) kJ·mol-1,要使該鍵斷裂并按方程式(8-26)發生反應至少需要600℃以上的高溫[150]。
(8-26)
(2) 反應機理。在不同的催化體系上N2O分解機理各不相同,但總結起來分為兩步:第一步N2O與活性中心相互作用造成N—O鍵斷裂,生成N2和吸附氧(方程式8-27),第二步是吸附氧的脫附[方程式(8-28)~(8-29)]。
(8-27)
(8-28)
(8-29)
8.6.2.3氧化亞氮的催化分解催化劑
自從Iwamoto等發現Cu-MFI對分解NOx有很高的活性后,一系列離子交換分子篩也被用來研究催化N2O直接分解[151]。從20世紀90年代開始,隨著對N2O認識的加深及對環境問題的日益重視,研究者開始致力于研究有較高催化N2O分解活性的催化劑,一些性能優異、有工業應用潛力的催化劑體系(如類水滑石分解產物催化劑及尖晶石催化劑等)被相繼研究出來。
N2O分解催化劑可大致分為金屬催化劑,氧化物催化劑和分子篩催化劑等。
(1) N2O的催化分解金屬催化劑。可以催化N2O直接分解的金屬催化劑主要是負載型貴金屬催化劑,如Pt、Pd、Rh、Ru、Au等單金屬催化劑,及Ag-Rh/Al2O3、Ag-Pd/Al2O3等雙金屬催化劑。對于單金屬催化劑,其催化活性的順序依次為Rh>Au>Ru>Pd>Pt;雙金屬Ag-M催化劑,主要活性區間在300℃以上(如果沒有特別說明,活性區間或活性溫度一般指沒有其他共存氣體且N2O轉化率達到50%以上的溫度區間)。而Rh、Ru、Au催化劑的活性區間為(200~400)℃,表現出較好的低溫催化活性。
(2) 氧化物催化劑。氧化物催化劑是目前研究最集中的可以催化N2O直接分解的催化體系。早期的研究主要在純氧化物催化劑和鈣鈦礦催化劑上展開,但它們對N2O分解反應的低溫催化活性都不盡人意。盡管如此,相關的機理研究為后繼研究者提供了很好的參考。近十年來,對于催化N2O直接分解具有高活性的復合氧化物催化劑如類水滑石分解產物及尖晶石催化劑等陸續被開發出來。

圖8-19 純氧化物催化N2O分解速率N2O分壓10 kPa,O2分壓0.1 kPa

圖8-20 LaMO3和La2MO4型復合氧化物上催化N2O直接分解活性的比較

圖8-21 Ni0.74Co0.26Co2O4和Co3O4催化N2O分解為N2和O2的活性氣體組成:(◆,◇)1000×10-6N2O;(■,□)1000×10-6N2O+10%O2;(△,▲)1000×10-6N2O+5%H2O;(○,●)1000×10-6N2O+10%O2+5%H2ONi0.74Co0.26Co2O4(實心符號),Co3O4(空心符號)[158]
(3) 分子篩催化劑。分子篩是由TO4四面體之間通過共享頂點而形成的三維四連接骨架。TO4四面體通過共享氧原子按照不同的連接方式最終形成多種孔道結構。特殊的孔道結構和骨架特征使其在成為催化劑時,具有特殊性質,如高的比表面積、擇形催化與分離。自20世紀60年代初美國聯合碳化物公司將A型沸石基催化劑應用于石油裂解以來,整個石油煉制的面貌發生改變。與此同時,分子篩催化劑在其他工業催化上的應用逐漸增多,相關理論不斷成熟,催化與吸附分離應用領域得到較大發展,有關詳細情況可參考相關專著[159]。
8.6.3氯氟烴的無害化
8.6.3.1氯氟烴的來源、危害和消除對策
氯氟烴類物質(Chlorofluorocarbons,CFCs)是一類分子中含氯和氟元素的碳氫化合物。CFCs都是無色、無味、無毒、無腐蝕性的氣體,化學性質十分穩定,當CFCs被釋放到大氣時,在低空的對流層中不易分解,然而當上升至平流層后,在紫外線照射下,會釋放出原子氯(強還原劑),這些原子氯與平流層中臭氧(強氧化劑)發生相互作用后,臭氧被還原成氧分子,從而減少了平流層中的臭氧含量,因此,臭氧層遭到破壞。
將CFCs無害化的方法可粗略的分為直接分解、催化分解(或光催化分解)以及加氫脫氯三大類。下面分別對CFCs無害化的催化分解、光催化分解以及加氫脫氯反應的特點和所用催化劑進行總結。
8.6.3.2氯氟烴的催化分解

8.6.3.3氯氟烴的光催化分解
目前,TiO2光催化理論和應用研究得到廣泛重視,研究表明,利用納米TiO2上的光催化反應可有效分解和去除水與空氣中的有機污染物。CFCs在大氣同溫層能夠被紫外光分解,因此,可以利用光催化劑在地面上讓CFCs加速分解。近幾年,CFCs的光催化分解成為重要的研究課題之一。
8.6.3.4氯氟烴的催化氫化脫氯無害化
在大多數研究報道中,用于CFCs的氫化脫氯反應催化劑為負載型貴金屬催化劑,如Pd、Pt和Rh,其中Pd催化劑因具有最高的轉化率和選擇性而得到深入研究。除了催化劑活性組分外,許多研究者也深入研究了催化劑載體在該類反應中的重要作用。對CFCs的氫化脫氯反應而言,催化劑載體一般為活性炭、氧化鋁或硫酸化的氧化鋁。
8.6.4羰基硫的催化水解和氧化
8.6.4.1羰基硫的環境效應
SSA對極地臭氧耗損有重要影響[161]。在極地的冬季,HNO3和H2O可凝結在SSA表面,從而清除了Cl自由基的清除劑NOx,而間接促進平流層臭氧耗損。此外,硫酸鹽氣溶膠還為HCl和ClONO2相對惰性的含氯物種重新分解為活性Cl自由基提供了催化反應的界面[162],而Cl自由基是臭氧耗損的關鍵物種之一。因此,COS是臭氧的間接耗損物質。
8.6.4.2羰基硫的催化水解和氧化
COS的脫除技術主要包括燃燒法、有機胺吸收法、催化水解法、氧化轉化法及加氫轉化法等[163]。其中,催化水解是目前脫除尾氣中COS的主流技術[164]。本節將主要介紹COS催化水解的研究進展。
COS催化水解的反應式為:
(8-30)
(1) COS水解催化劑體系。催化劑活性組分有堿金屬、堿土金屬、過渡金屬氧化物以及稀土金屬硫氧化物;載體主要有γ-Al2O3、TiO2和活性炭。由于成本和應用歷史原因,γ-Al2O3仍是市場上COS催化水解的主要催化劑載體。事實上,γ-Al2O3本身也具有一定的催化水解活性[165]。例如,在無氧體系中,空速2 500 h-1和溫度20℃的條件下,γ-Al2O3對COS的穩態轉化率達54.9%[165],當空速增加到12 300 h-1時,γ-Al2O3對COS的穩態轉化率仍可保持約20%[166]。
(2) COS催化水解動力學。研究發現,COS在γ-Al2O3上的催化水解反應滿足Langmiur-Hinshelwood動力學模型。其反應速率可表達為:

(8-31)
式中,r0為初始反應速率;k1為表面反應速率常數;XS為催化劑表面活性位濃度;K為吸附平衡常數。
(3) COS催化反應機理。COS首先與催化劑表面的堿性羥基作用生成THC,THC可在質子酸的作用下分解生成CO2和H2S,也可在表面吸附水的參與下分解為CO2和H2S。該反應機理與George接近,也與動力學研究中得到的Langmiur-Hinshelwood模型相吻合。Akimoto M等[167]認為,COS的催化水解還與催化劑表面還原位點和L酸位相關。當反應氣氛中加入戊烯后,由于其在L酸位與H2O發生競爭吸附,而使催化活性降低;而反應體系中加入SO2后,Akimoto M認為SO2與COS在催化劑表面的還原性位點的競爭吸附導致催化活性的降低。Liu J F等[165]利用原位紅外光譜研究發現,如圖8-22所示COS在催化劑表面吸附的同時伴隨著表面堿羥基(B堿)的消耗。因此,COS在還原性位點發生吸附值得商榷。

圖8-22 γ-Al2O3含氧體系催化水解COS的原位紅外光譜φ(COS)=0.05%,φ(O2)=95%,298 K
(4) COS催化氧化。COS的催化氧化研究相對較少。奚強等[168]利用金屬酞菁(TsPc)在液相中可將COS催化氧化為單質硫。不同金屬酞菁的活性順序為CoTsPc>ZnTsPc>NiTsPc>FeTsPc>MnTsPc>CuTsPc。COS先被水解為HS-,HS-進一步被氧化為S。在多相催化中,李福林等[169]開發了一步法羰基硫脫硫劑,脫硫劑含質量分數1%~20%的Al2O3、TiO2、ZrO2和CuO中的一種金屬氧化物,質量分數4%~12%的Na2CO3、K2CO3、NaOH和KOH中的一種或幾種為調變劑,質量分數0.01%~0.1%的磷酸鹽、磺酸鹽和醇胺為傳質促進劑,該催化氧化劑可在60℃有氧條件下,將COS催化氧化為單質硫。
參考文獻:
[1]吳越.催化化學[M].北京:科學出版社,2000.
[2]李燦,林勵吾.催化基礎國家重點實驗室[J].中國基礎科學,2005,(2):30-32.
[3]Ertl G,Kn?zinger H,Weitkamp J.Environmental catalysis[M].Weinheim:Wiley-VCH,1999.
[4]Grassian V H.Environmental catalysis[M].London:Taylor & Francis Group,2005.
[5]巖本正和.環境觸媒ハンドブック[M].東京:ヌ·ティー·エス,2001.
[6]Janssen F,Van Santen R A.Environmental catalysis[M].London:Imperial College Press,1999.
[7]Fukuoka A,Dhepe P L.Catalytic conversion of cellulose into sugar alcohols[J].Angewandte Chemie International Edition,2006,45:5161-5163.
[8]Luo C,Wang S,Liu H C.Cellulose conversion into polyols catalyzed by reversibly formed acids and supported ruthenium clusters in hot water[J].Angewandte Chemie International Edition,2007,46:7636-7639.
[9]http://www.catalystgrp.com/catalystsandchemicals.html.
[10]http://www.catalystgrp.com/newsandpress2.html#Anchor-Chemi-48622.Catalysts:possible changes on the horizon.Chemical Week,April,12-19,2006.
[11]Orilik S N.Contemporary problems in the selective catalytic reduction of nitrogen oxides(NOx)[J].Theoretical and Experimental Chemistry,2001,37(3):135-162.
[12]Heck R M,Gulati S,Farrauto R J.The application of monoliths for gas phase catalytic reactions[J].Chemical Engineering Journal,2001,82:149-156.
[13]Farrauto R J,Heck R M.Catalytic converters:state of the art and perspective[J].Catalysis Today,1999,51:351-360.
[14]Meunier F C,Ross J R H.Effect of ex situ treatments with SO2on the activity of a low loading silver-alumina catalyst for the selective reduction of NO and NO2by propene[J].Applied Catalysis B:Environmental,2000,24(1):23-32.
[15]Larese C,Granados M L,Galistro F C,et al.TWC deactivation by lead:a study of the RN-CeO2system[J].Applied Catalysis B:Environmental,2006,62:132-143.
[16]Wachs I E,Deo G,Weckhuysen B M,et al.Selective catalytic reduction of NO with NH3over supported vanadia catalysts[J].Journal of Catalysis,1996,161:211-221.
[17]Li J,Chang H,Ma L,et al.Low-temperature selective catalytic reduction of NOx with NH3over metal oxide and zeolite catalysts——A review[J].Catalysis Today,2011,175:147-156.
[18]Ka?par J,Fornasiero P,Hickey N.Automotive catalytic converts:current status and some perspectives[J].Catalysis Today,2003,77:410-449.
[19]Heck R M,Fattauto R J.Automobile exhaust catalysts[J].Applied Catalysis A:General,2001,221:443-457.
[20]Oh S H,Fisher G B,Carpenter J E,et al.Comparative kinetic studies of CO-O2and CO-NO reactions over single crystal and supported rhodium catalysts[J].Journal of Catalysis,1986,100(2):360-376.
[21]Twigg M V.Twenty-five years of autocatalysts[J].Platinum Metals Review,1999,43:168-171.
[22]http://en.wikipedia.org/wiki/Oxygen_sensor.
[23]Ka?par J,Fornasiero P,Hickey N.Automotive catalytic converters:current status and some perspectives[J].Catalysis Today,2003,77:419-449.
[24]Heck R M,Fattauto R J.Automobile exhaust catalysts[J].Applied Catalysis A:General,2001,221:443-457.
[25]Hu Z,Wan C Z,Lui Y K,et al.Design of a novel Pd three-way catalyst:integration of catalytic functions in three dimensions[J].Catalysis Today,1996,30:83-89.
[26]Oh S H,Fisher G B,Carpenter J E,et al.Comparative kinetic studies of CO-O2and CO-NO reactions over single crystal and supported rhodium catalysts[J].Journal of Catalysis,1986,100:360-376.
[27]Taylor K C,Schlatter J C.Selective reduction of nitric-oxide over noble metals[J].Journal of Catalysis,1980,63:53-71.
[28]Ka?par J,Fornasiero P,Graziani M.Use of CeO2-based oxides in the three-way catalysis[J].Catalysis Today,1999,50:285-298.
[29]Webster D E.25 years of catalytic automotive pollution control:a collaborative effort[J].Top Catal,2001,16/17:33-38.
[30]Johnson T V.Diesel emission control technology-2003 in review[J].SAE paper,2004-01-0070.
[31]Koebel M,Elsener M,Madia G.Recent advances in the development of urea-SCR for automotive applications[J].SAE paper,2001-01-3625.
[32]Koebel M,Elsener M,Kleemann M.Urea-SCR:a promising technique to reduce NOx emissions from automotive diesel engines[J].Catalysis Today,2000,59:335-345.
[33]Liu F,He H,Zhang C.Novel iron titanate catalyst for the selective catalytic reduction of NO with NH3in the medium temperature range[J].Chemical Communications,2008,2043-2045.
[34]Yang S,Li J,Wang C,et al.Fe-Ti spinel for the selective catalytic reduction of NO with NH3:mechanism and structure-activity relationship[J].Applied Catalysis B:Environmental,2012,117-118:73-80.
[35]Shan W P,Liu F D,He H,et al.The remarkable improvement of a Ce-Ti based catalyst for NOx abatement[J].ChemCatChem,2011,3:1286-1289.
[36]Shan W P,Liu F D,He H,et al.Novel cerium-tungsten mixed oxide catalyst for the selective catalytic reduction of NOx with NH3[J].Chemical Communications,2011,47:8046-8048.
[37]Kwak J H,Tonkyn R G,Kim D H,et al.Excellent activity and selectivity of Cu-SSZ-13 in the selective catalytic reduction of NOx with NH3[J].Journal of Catalysis,2010,275:187-190.
[38]Xie L,Liu F,Ren L,et al.Excellent performance of one-pot synthesized Cu-SSZ-13 catalyst for the selective catalytic reduction of NO with NH3[J].Environmental Science & Technology,2014,48:566-572.
[39]Cavataio G,Girard J,Patterson J E,et al.Laboratory testing of urea-SCR formulations to meet tier 2 bin 5 emissions[J].SAE paper,2007-01-1575.
[40]Sato S,Yuu Y,Yahiro H,et al.Cu-ZSM-5 zeolite as highly active catalyst for removal of nitrogen monoxide from emission of diesel engines[J].Applied Catalysis,1991,70:1-5.
[41]Burch R,Breen J P,Meunier F C.A review of the selective reduction of NOx with hydrocarbons under lean-burn conditions with non-zeolitic oxide and platinum group metal catalysts[J].Applied Catalysis B:Environmental,2002,39:283-303.
[42]He H,Yu Y B.Selective catalytic reduction of NOx over Ag/Al2O3catalyst:from reaction mechanism to diesel engine test[J].Catalysis Today,2005,100:37-47.
[43]Li J H,Zhu Y Q,Ke R,et al.Improvement of catalytic activity and sulfur-resistance of Ag/TiO2-Al2O3for NO reduction with propene under lean burn conditions[J].Applied Catalysis B:Environmental,2008,80:202-213.
[44]Houel V,Millington P,Rajaram R,et al.Fuel effects on the activity of silver hydrocarbon-SCR catalysts[J].Applied Catalysis B:Environmental,2007,73:203.
[45]Yu Y B,He H,Feng Q C.Novel enolic surface species formed during partial oxidation of CH3CHO,C2H5OH and C3H6on Ag/Al2O3:an in situ DRIFTS study[J].Journal of Physical Chemistry B,2003,107:13090-13092.
[46]Yu Y B,He H,Feng Q C,et al.Mechanism of the selective catalytic reduction of NOx by C2H5OH over Ag/Al2O3[J].Applied Catalysis B:Environmental,2004,49:159-171.
[47]Taatjes C A,Hansen N,McIlroy A,et al.Enols are common intermediates in hydrocarbon oxidation[J].Science,2005,308:1887-1889.
[48]Liu F D,Yu Y B,He H.Environmentally-benign catalysts for the selective catalytic reduction of NOx from diesel engines:structure-activity relationship and reaction mechanism aspects[J].Chemical Communications,2014,50:8445-8463.
[49]Yu Y B,He H,Zhang X L,et al.A common feature of H2-assisted HC-SCR over Ag/Al2O3[J].Catalysis Science & Technology,2014,4:1239-1245.
[50]Miyoshi N,Matsumoto S,Katon K,et al.Development of new concept three-way catalyst for automotive lean-burn engines[J].SAE paper,950809-950821.
[51]歐翔飛,羅東曉.國內壓縮天然氣汽車產業發展分析[J].天然氣工業,2007,27:129-132.
[52]彭紅濤.天然氣汽車發展現狀及對策[J].汽車工業研究,2006,1:47-48.
[53]Hansen A C,Zhang Q,Lyne P W L.Ethanol-diesel fuel blends——A review[J].Bioresource Technology,2005,96:277-285.
[54]Wheals A E,Basso L C,Alves D M G,et al.Fuel ethanol after 25 years[J].Trends in Biotechnology,1999,17:482-487.
[55]Choudhary T V,Banerjee S,Choudhary V R.Catalysts for combustion of methane and lower alkanes[J].Applied Catalysis A:General,2002,234:1-23.
[56]Li Y J,Armor J N.Catalytic reduction of nitrogen oxides with methane in the presence of excess oxygen[J].Applied Catalysis B:Environmental,1992,1:L31-L40.
[57]新井紀男.燃燒生成物的發生與抑制技術[M].北京:科學出版社,2001:77-94.
[58]Seyedeyn Azad F,Zhang D K.Selective catalytic reduction of nitric oxide over Cu and Co ion-exchanged ZSM-5 zeolite:the effect of SiO2/Al2O3ratio and cation loading[J].Catalysis Today,2001,68:161-171.
[59]Wang X,Chen H,Sachtler W M H.Selective reduction of NOx with hydrocarbons over Co/MFI prepared by sublimation of CoBr2and other methods[J].Applied Catalysis B:Environmental,2001,29:47-60.
[60]Long R Q,Yang R T.Selective catalytic reduction of NO with ammonia over Fe3+-exchanged mordenite(Fe-MOR):catalytic performance,characterization,and mechanistic study[J].Journal of Catalysis,2002,207:274-285.
[61]Wang L,Qin G S,Weng D.Location and nature of Cu species in Cu/SAPO-34 for selective catalytic reduction of NO with NH3[J].Journal of Catalysis,2012,289:21-29.
[62]Nikolopoulos A A,Stergioula E S,Efthimiadis E A,et al.Selective catalytic reduction of NO by propene in excess oxygen on Pt- and Rh-supported alumina catalysts[J].Catalysis Today,1999,54:439-450.
[63]Denton P,Giroir-Fendler A,Schuurman Y,et al.A redox pathway for selective NOx reduction:stationary and transient experiments performed on a supported Pt catalyst[J].Applied Catalysis A:General,2001,220:141-152.
[64]Long R Q,Yang R T.Temperature-programed desorption/surface reaction(TPD/TPSR) study of Fe-exchanged ZSM-5 for selective catalytic reduction of nitric oxide by ammonia[J].Journal of Catalysis,2001,198:20-28.
[65]Svachula J,Alemany L J,Ferlazzo N,et al.Oxidation of SO2to SO3over honeycomb deNOxing catalysts[J].Industrial & Engineering Chemistry Research,1993,32:826-834.
[66]Orlik S N,Ostapyuk V A,Martsenyukkukharuk M G.Selective reduction of nitrogen-oxides with ammonia on V2O5/TiO2catalysts[J].Kinet Catal,1995,36:284-289.
[67]Inomata M,Miyamoto A,Murakami Y.Mechanism of the reaction of NO and NH3on vanadium oxide catalyst in the presence of oxygen under the dilute gas condition[J].Journal of Catalysis,1980,62:140-148.
[68]Pinoy L J,Hosten L H.Experimental and kinetic modeling study of deNOx on an industrial V2O5-WO3/TiO2catalyst[J].Catalysis Today,1993,17:151-158.
[69]宣小平,姚強,岳長濤,等.選擇性催化還原法脫硝研究進展[J].煤炭轉化,2002,25:26-31.
[70]Odenbrand C U I,Lundin S T,Andersson L A H.Catalytic reduction of nitrogen oxides Ⅰ.The reduction of NO[J].Applied Catalysis,1985,18:335-352.
[71]Boulahouache A,Kons G,Lintz H G,et al.Oxidation of carbon monoxide on platinum-tin dioxide catalysts at low temperatures[J].Applied Catalysis A:General,1992,91:115-123.
[72]Komatsu T,Uddin M A,Yashima T.Zeolites,A refined tool for designing catalytic sites zeolites[M].Amsterdam:Elsevier,1995:437.
[73]Willey R J,Eldridge J W,Kittrell J R.Mechanistic model of the selective catalytic reduction of nitric oxide with ammonia[J].Ind Eng Chem Pro Res Dev,1985,24:226-233.
[74]東方鍋爐(集團)股份有限公司環保工程公司脫氮技術交流資料. 2004.
[75]陳杭君,趙華,丁經緯.火電廠煙氣脫硝技術介紹[J].熱力發電,2005,2:15-18.
[76]劉今.發電廠煙氣脫硝技術——SCR法[J].江蘇電機工程,1996,15(1):51-55.
[77]Haas L A,Khalafalla S E.Kinetic evidence of a reactive intermediate in reduction of SO2with CO[J].Journal of Catalysis,1973,29:264-269.
[78]Happel J,Hnatow M A,Bajars L,et al.Lanthanum titanate catalyst-sulfur dioxide reduction[J].Industrial & Engineering Chemistry Research,1975,14:154-158.
[79]Tschope A,Liu W,Ying J Y.Redox activity of nonstoichiometric oxide-based nanocrystalline catalyst[J].Journal of Catalysis,1995,157:42-50.
[80]Yeh T,Demeki R T,Strakey J P.Combined SO2/NOx removal from flue gases[J].Environmental Progress & Sustainable Energy,1985,4:223-229.
[81]Yoo J S,Bhattacharyya A A,Radlowski C A.Advanced De-SOx catalyst:mixed solid solution spinels with cerium oxide[J].Applied Catalysis B:Environmental,1992,1:169-189.
[82]Klepeis N E,Nelson W C,Ott W R,et al.The National human activity pattern survey (NHAPS):a resource for assessing exposure to environmental pollutants[J].Journal of Exposure Analysis and Environment Epidemiology,2001,11:231-252.
[83]朱天樂.室內空氣污染控制[M].北京:化學工業出版社,2002.
[84]Fujishima A,Hond K.Electrochemical photolysis of water at a semiconductor electrode[J].Nature,1972,238:37-38.
[85]張金龍,陳峰,何斌.光催化[M].上海:華東理工大學出版社,2004.
[86]Yu C,Crump D.A review of the emission of VOCs from polymeric materials used in buildings[J].Building and Environment,1998,33:357-374.
[87]Molhavc L.Total volatile organic compounds (TVOC) in indoor air quality investigations[J].Indoor Air,1997,7:225-240.
[88]Jones A P.Indoor air quality and health[J].Atmospheric Environment,1999,33:4535-4564.
[89]Sano T,Negishi N,Uchino K,et al.Photocatalytic degradation of gaseous acetaldehyde on TiO2with photo deposited metals and metal oxides[J].Journal of Photochemistry and Photobiology A:Chemistry,2003,160:93-98.
[90]Sinha A K,Suzuki K.Preparation and characterization of novel mesoporous ceria-titania[J].Journal of Physical Chemistry B,2005,109:1708-1714.
[91]Haruta M,Yamada N,Kobayashi T,et al.Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon monoxide[J].Journal of Catalysis,1989,115:301-309.
[92]Sekine Y.Oxidative decomposition of formaldehyde by metal oxides at room temperature[J].Atmospheric Environment,2002,36:5543-5547.
[93]Zhang C B,He H,Tanaka K.Catalytic performance and mechanism of a Pt/TiO2catalyst for oxidation of formaldehyde at room temperature[J].Applied Catalysis B:Environmental,2006,65:37-43.
[94]Francke K P,Miessner H,Rudolph R.Plasmacatalytic processes for environmental problems[J].Catalysis Today,2000,59:411-416.
[95]Yamamoto T,Hill C.Methods and apparatus for controlling toxic compounds using catalysis-assisted non-thermal plasma:US,5609736[P].1997.
[96]Futsmura S,Zhang A,Einaga H,et al.Involvement of catalyst materials in nonthermal plasma chemical processing of hazardous air pollutants[J].Catalysis Today,2002,72:259-265.
[97]Subrahmanyam C H,Magureanu M,Renken A,et al.Catalytic abatement of volatile organic compounds assisted by non-thermal plasma,Part 1.A novel dielectric barrier discharge reactor containing catalytic electrode[J].Applied Catalysis B:Environmental,2006,65:150-156.
[98]Jacoby W A,Maness P C,Wolfrum E J,et al.Mineralization of bacterial cell mass on a photocatalytic surface in air[J].Environmental Science & Technology,1998,32:2650-2653.
[99]金宗哲.無機抗菌材料及其應用[M].北京:化學工業出版社,2004.
[100]Thurman R B,Gerba C P.The molecular mechanisms of copper and silver ion disinfection of bacteria and viruses[J].CRC Critical Reviews in Enviromental Control,1989,18:295-315.
[101]Feng Q L,Wu J,Chen G Q,et al.A mechanistic study of the antibacterial effect of silver ions on Escherichia coli and Staphylococcus aureus[J].Journal of Biomedical Materials Research,2000,52:662-668.
[102]Inoue Y,Hoshino M,Takahashi H,et al.Bactericidal activity of Ag-zeolite mediated by reactive oxygen species under aerated conditions[J].Journal of Inorganic Biochemistry,2002,92:37-42.
[103]Pape H L.Solano-Serena F,Contini P,et al.Involvement of reactive oxygen species in the bactericidal activity of activated carbon fibre supporting silver Bactericidal activity of ACF (Ag) mediated by ROS[J].Journal of Inorganic Biochemistry,2004,98:1054-1060.
[104]Ruan Q J,Zhang W D.Tunable morphology of Bi2Fe4O9crystals for photocatalytic oxidation[J].Journal of Physical Chemistry C,2009,113:4168-4173.
[105]Hu C,Peng T,Hu X,et al.Plasmon-induced photodegradation of toxic pollutants with Ag-AgI/Al2O3under visible-light irradiation[J].Journal of the American Chemical Society,2010,132:857-862.
[106]Zhou X F,Hu C,Hu X X,et al.Enhanced electron transfer and silver-releasing suppression in Ag-AgBr/titanium-doped Al2O3suspensions with visible-light irradiation[J].Journal of Hazardous Materials,2012,219:276-282.
[107]Wang Q,Zhang M,Chen C,et al.Photocatalytic aerobic oxidation of alcohols on TiO2:the acceleration effect of a Bronsted acid[J].Angewandte Chemie International Edition,2010,49:7976-7979.
[108]Li Y,Wen B,Ma W,et al.Photocatalytic degradation of aromatic pollutants:a pivotal role of conduction band electron in distribution of hydroxylated intermediates[J].Environmental Science & Technology,2012,46:5093-5099.
[109]Zhao Y,Ma W,Li Y,et al.The surface-structure sensitivity of dioxygen activation in the anatase-photocatalyzed oxidation reaction[J].Angewandte Chemie International Edition,2012,51:3188-3192.
[110]Fenton H J H.Oxidation of tartaric acid in the presence of iron[J].Journal of the Chemical Society,1894,65:899-901.
[111]Eisenhauer H R.Oxidation of phenolic wastes[J].Journal of the Water Pollution Control Federation,1964,36:1116-1128.
[112]Martins R C,Amaral-Silva N,Quinta-Ferreira R M.Ceria based solid catalysts for Fenton’s depuration of phenolic wastewaters,biodegradability enhancement and toxicity removal[J].Applied Catalysis B:Environmental,2010,99:135-144.
[113]Bremner D H,Burgess A E,Houllemare D,et al.Phenol degradation using hydroxyl radicals generated from zero-valent iron and hydrogen peroxide[J].Applied Catalysis B:Environmental,2006,63:15-19.
[114]Segura Y,Martínez F,Melero J A,et al.Enhancement of the advanced Fenton process (Fe0/H2O2) by ultrasound for the mineralization of phenol[J].Applied Catalysis B:Environmental,2012,113-114:100-106.
[115]Guimaraes I R,Giroto A,Oliveira L C A,et al.Synthesis and thermal treatment of Cu-doped goethite:oxidation of quinoline through heterogeneous Fenton process[J].Applied Catalysis B:Environmental,2009,91:581-586.
[116]Nie Y,Hu C,Qu J,et al.Photoassisted degradation of endocrine disruptors over CuOx-FeOOH with H2O2at neutral pH[J].Applied Catalysis B:Environmental,2009,87:30-36.
[117]Zhang L,Nie Y,Hu C,et al.Enhanced Fenton degradation of Rhodamine B over nanoscaled Cu-doped LaTiO3perovskite[J].Applied Catalysis B:Environmental,2012,125:418-424.
[118]Navalon S,Miguel M,Martin R,et al.Enhancement of the catalytic activity of supported gold nanoparticles for the Fenton reaction by light[J].Journal of the American Chemical Society,2011,133:2218-2226.
[119]Xu L,Wang J.Magnetic nanoscaled Fe3O4/CeO2composite as an efficient Fenton-like heterogeneous catalyst for degradation of 4-chlorophenol[J].Environmental Science & Technology,2012,46:10145-10153.
[120]彭長征.飲用水的臭氧氧化技術[J].山西建筑,2006,32:173-174.
[121]Nawrocki J,Kasprzyk-Hordern B.The efficiency and mechanisms of catalytic ozonation[J].Applied Catalysis B:Environmental,2010,99:27-42.
[122]Lü A,Hu C,Nie Y,et al.Catalytic ozonation of toxic pollutants over magnetic cobalt and manganese co-doped γ-Fe2O3[J].Applied Catalysis B:Environmental,2010,100:62-67.
[123]Lü A,Hu C,Nie Y,et al.Catalytic ozonation of toxic pollutants over magnetic cobalt-doped Fe3O4suspensions[J].Applied Catalysis B:Environmental,2012,117:246-252.
[124]Yang L,Hu C,Nie Y,et al.Catalytic ozonation of selected pharmaceuticals over mesoporous alumina-supported manganese oxide[J].Environmental Science & Technology,2009,43:2525-2529.
[125]Yang L,Hu C,Nie Y,et al.Surface acidity and reactivity of β-FeOOH/Al2O3for pharmaceuticals degradation with ozone:In situ ATR-FTIR studies[J].Applied Catalysis B:Environmental,2010,97:340-346.
[126]Zhao L,Sun Z,Ma J.Novel relationship between hydroxyl radical initiation and surface group of ceramic honeycomb supported metals for the catalytic ozonation of nitrobenzene in aqueous solution[J].Environmental Science & Technology,2009,43:4157-4163.
[127]Beltrán F J,Pocostales P,lvarez P M,et al.Catalysts to improve the abatement of sulfamethoxazole and the resulting organic carbon in water during ozonation[J].Applied Catalysis B:Environmental,2009,92:262-270.
[128]陳嵩,孫珮石,李福華,等.CWO技術處理我國高濃度工業廢水的應用研究[J].貴州環保科技,2003,9:1-5.
[129]Keav S,Monteros A E,Jr J B,et al.Wet air oxidation of phenol over Pt and Ru catalysts supported on cerium-based oxides:resistance to fouling and kinetic modelling[J].Applied Catalysis B:Environmental,2014,150-151:402-410.
[130]Ayusheev A B,Taran O P,Seryak I A,et al.Ruthenium nanoparticles supported on nitrogen-doped carbon nanofibers for the catalytic wet air oxidation of phenol[J].Applied Catalysis B:Environmental,2014,146:177-185.
[131]Arena F,Italiano C,Ferrante G D,et al.A mechanistic assessment of the wet air oxidation activity of MnCeOx catalyst toward toxic and refractory organic pollutants[J].Applied Catalysis B:Environmental,2014,144:292-299.
[132]Li Y,Xu Y,Chen X,et al.High catalytic activity of Mo-Zn-Al-O catalyst for dye degradation:effect of pH in the impregnation process[J].Applied Catalysis B:Environmental,2014,160-161:115-121.
[133]World Health Organization.Health hazards from nitrates in drinking water[C].Copenhagen:WHO Regional Office for Europe,1985.
[134]Drinking water regulation.Health advisories[M].Washington D C:Office of Water,1995.
[135]Zhao W,Zhu X,Wang Y,et al.Catalytic reduction of aqueous nitrates by metal supported catalysts on Al particles[J].Chemical Engineering Journal,2014,254:410-417.
[137]Kim M S,Lee D W,Chung S H,et al.Pd-Cu bimetallic catalysts supported on TiO2-CeO2mixed oxides for aqueous nitrate reduction by hydrogen[J].Journal of Molecular Catalysis A:Chemical,2014,392:308-314.
[138]Al-Fatesh A S,Ibrahim A A,Haider S,et al.Sustainable production of synthesis gases via state of the art metal supported catalytic systems:an overview[J].Journal of the Chinese Chemical Society,2013,60:1297-1308.
[139]Osako T,Watson E J,Dehestani A,et al.Methane oxidation by aqueous osmium tetroxide and sodium periodate:inhibition of methanol oxidation by methane[J].Angewandte Chemie International Edition,2006,45:7433-7436.
[140]Rostrupnielsen J R,Hansen J H B.CO2-Reforming of methane over transition-metals[J].Journal of Catalysis,1993,144:38-49.
[141]Liu H,Li Y,Wu H,et al.Promoting effect of glucose and beta-cyclodextrin on Ni dispersion of Ni/MCM-41 catalysts for carbon dioxide reforming of methane to syngas[J].Fuel,2014,136:19-24.
[142]Ashcroft A T,Cheetham A K,Green M L H,et al.Partial oxidation of methane to synthesis gas-using carbon-dioxide[J].Nature,1991,352:225-226.
[143]Estifaee P,Haghighi M,Babaluo A A,et al.The beneficial use of non-thermal plasma in synthesis of Ni/Al2O3-MgO nanocatalyst used in hydrogen production from reforming of CH4/CO2greenhouse gases[J].Journal of Power Sources,2014,257:364-373.
[144]Guo J J,Lou H,Zhao H,et al.Improvement of stability of out-layer MgAl2O4spinel for a Ni/MgAl2O4/Al2O3catalyst in dry reforming of methane[J].Reaction Kinetics and Catalysis Letters,2005,84:93-100.
[145]陶凱.甲烷二氧化碳重整催化劑制備及反應性能研究[D].大連:大連理工大學,2007.
[146]Lima S M,Assaf J M,Pena M A,et al.Structural features of La1-xCexNiO3mixed oxides and performance for the dry reforming of methane[J].Applied Catalysis A:General,2006,311:94-104.
[147]Topalidis A,Petrakis D E,Ladavos A,et al.A kinetic study of methane and carbon dioxide interconversion over 0.5%Pt/SrTiO3catalysts[J].Catalysis Today,2007,127:238-245.
[148]Bradford M C J,Vannice M A.CO2reforming of CH4[J].Catalysis Reviews,1999,41:1-42.
[149]Wei J M,Iglesia E.Isotopic and kinetic assessment of the mechanism of reactions of CH4with CO2or H2O to form synthesis gas and carbon on nickel catalysts[J].Journal of Catalysis,2004,224:370-383.
[150]Kapteijn F,Rodriguezmirasol J,Moulijn J A.Heterogeneous catalytic decomposition of nitrous oxide[J].Applied Catalysis B:Environmental,1996,9:25-64.
[151]Boron P,Chmielarz L,Gurgul J,et al.The influence of the preparation procedures on the catalytic activity of Fe-BEA zeolites in SCR of NO with ammonia and N2O decomposition[J].Catalysis Today,2014,235:210-225.
[152]Zabilskiy M,Erjavec B,Djinovic P,et al.Ordered mesoporous CuO-CeO2mixed oxides as an effective catalyst for N2O decomposition[J].Chemical Engineering Journal,2014,254:153-162.
[153]Haber J,Nattich M,Machej T.Alkali-metal promoted rhodium-on-alumina catalysts for nitrous oxide decomposition[J].Applied Catalysis B:Environmental,2008,77:278-283.
[154]Wang J,Yasuda H,Inumaru K,et al.Catalytic decomposition of dinitrogen oxide over perovskite-related mixed oxides[J].The Bulletin of the Chemical Society of Japan,1995,6:1226-1231.
[155]Christopher J,Swamy C S.Studies on the catalytic decomposition of N2O on LnSrFeO4(Ln=La,Pr,Nd,Sm and Gd)[J].Journal of Molecular Catalysis A:Chemical,1991,68:199-213.
[156]王立秋,張守臣,劉長厚.類水滑石復合產物催化消除氮氧化物的研究進展[J].化工進展,2003,10:1076-1080.
[157]Kannan S,Swamy C S.Catalytic decomposition of nitrous oxide over calcined cobalt aluminum hydrotalcites[J].Catalysis Today,1999,53:725-737.
[158]Yan L,Ren T,Wang X L,et al.Catalytic decomposition of N2O over MxCo1-xCo2O4(M=Ni,Mg) spinel oxides[J].Applied Catalysis B:Environmental,2003,45:85-90.
[159]Xie P,Ma Z,Zhou H,et al.Catalytic decomposition of N2O over Cu-ZSM-11 catalysts[J].Microporous and Mesoporous Materials,2014,191:112-117.
[160]Fung S C,Sinfelt J H.Hydrogenolysis of methyl-chloride on metals[J].Journal of Catalysis,1987,103:220-223.
[161]Andreae M O,Crutzen P J.Atmospheric aerosols:biogeochemical sources and role in atmospheric chemistry[J].Science,1997,276:1052-1058.
[162]Leung F Y T.Elucidation of the origins of stratospheric sulfate aerosols by isotopic methods[D].California Institute of Technology,2003.
[163]Rhodes C,Riddel S A,West J,et al.The low-temperature hydrolysis of carbonyl sulfide and carbon disulfide:a review[J].Catalysis Today,2000,59:443-464.
[164]Wang H,Yi H,Tang X,et al.Catalytic hydrolysis of COS over calcined CoNiAl hydrotalcite-like compounds modified by cerium[J].Applied Clay Science,2012,70:8-13.
[165]Liu J F,Liu Y C,Xue L,et al.Oxygen poisoning mechanism of catalytic hydrolysis of COS over Al2O3at room temperature[J].Acta Physico-Chimica Sinica,2007,23:997-1002.
[166]Thomas B,Williams B P,Young N,et al.Ambient temperature hydrolysis of carbonyl sulfide using gamma-alumina catalysts:effect of calcination temperature and alkali doping[J].Catalysis Letters,2003,86:201-205.
[167]Akimoto M,Lana I G D.Role of reduction sites in vapor-phase hydrolysis of carbonyl sulfide over alumina catalysts[J].Journal of Catalysis,1980,62:84-93.
[168]奚強,劉常坤,趙春芳,等.酞菁鈷液相催化氧化羰基硫(COS)的研究[J].離子交換與吸附,1997,(6):603-607.
[169]李福林,王樹東,吳迪鏞,等.一步法羰基硫脫硫劑:中國,CN1340373[P].2002-04-11.
現代催化化學講座
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
