皮膚異色病樣淀粉樣變病二例
2016-08-13 08:58:59林爾藝吳鐵強顧有守
中國麻風皮膚病雜志 2016年7期
艾 菁 林爾藝 吳鐵強 顧有守 楊 斌
?
皮膚異色病樣淀粉樣變病二例
艾 菁 林爾藝 吳鐵強 顧有守 楊 斌
患者1,男,49歲,病程9年;患者2,女,21歲,病程3年。兩例患者均表現為全身彌漫性色素沉著夾雜點狀白斑,伴苔蘚樣丘疹,偶有水皰。家族中無類似病史。組織病理示:真皮乳頭層淀粉樣物質沉積。
淀粉樣變; 皮膚異色病
皮膚異色病樣淀粉樣變病(Poikiloderma-like Cutaneous Amyloidosis,簡稱PCA綜合征)是原發性皮膚淀粉樣變病的一個亞型,臨床上較為少見,現將我院近期診治的兩例患者報道如下。
臨床資料 患者1,男,49歲。因全身褐色色素沉著、色素減退相間伴苔蘚樣丘疹9年,水皰1年就診。患者9年前無明顯誘因于項部、腹部及四肢出現暗紅色斑疹及丘疹,伴輕度瘙癢,日曬后加重,未予任何處理,皮損逐漸擴大并變為黑褐色,夾雜點狀白斑,曾多次至各大醫院就診(具體治療不詳),療效不佳。近一年來,患者癥狀加重,并反復于皮疹上出現綠豆大水皰,經數天后水皰可自行消退。既往史、家族史無特殊。
體檢:各系統檢查無異常。皮膚科檢查:項部可見對稱性黑褐色斑疹,略高于皮膚,融合形成波紋狀外觀。雙上肢及腹部可見大片網狀色素沉著斑,夾雜點狀白斑,散在針尖大丘疹,輕度萎縮,毛細血管擴張,前臂可見數個綠豆大水皰,皰壁緊張,皰液清,尼氏征陰性。膝部可見黑褐色斑丘疹,融合成片,表面粗糙,小腿脛前可見黃豆大黑褐色丘疹,呈串珠樣排列(圖1、2)。
實驗室檢查:血常規、肝腎功能均正常。類天皰瘡抗體:陰性。組織病理學檢查示:(1)水皰區表皮內皰見纖維蛋白,真皮乳頭見嗜伊紅均質物沉積,散在噬色素細胞,血管周圍少量淋巴細胞浸潤;(2)斑疹區表皮角化過度,基底細胞液化變性,色素失禁,真皮淺層散在噬黑素細胞,見嗜伊紅均質物沉積,血管周圍少許淋巴細胞(圖3、4)。甲基紫染色:陽性(圖5);免疫熒光:陰性。結合臨床診斷:皮膚異色病樣淀粉樣變病。
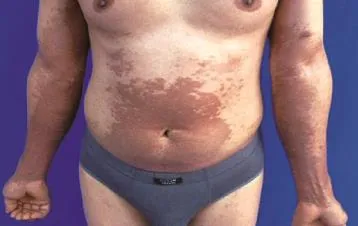
圖1 雙上肢及腹部大片網狀色素沉著斑,夾雜點狀白斑,散在針尖大丘疹,輕度萎縮,毛細血管擴張
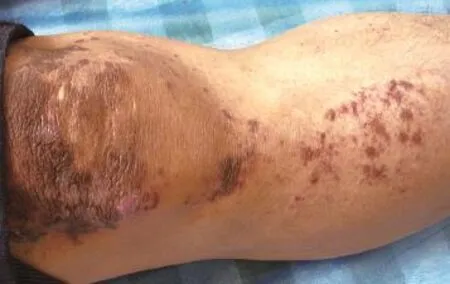
圖2 膝部可見黑褐色斑丘疹,融合成片,表面粗糙,小腿脛前可見黃豆大黑褐色丘疹,呈串珠樣排列
患者2,女,21歲。因軀干、四肢褐色斑片、水皰伴瘙癢3年就診。患者3年前無明顯誘因于四肢及背部出現黑褐色斑片,夾雜點狀白斑,針尖大水皰,伴瘙癢,搔抓后水皰可破裂,遺留褐色小丘疹,日曬后加重,3年來癥狀逐漸加重,遂來我院就診。既往史、家族史無特殊。
體檢:各系統檢查無異常。皮膚科檢查:背部及四肢可見彌漫性網狀色素沉著斑,夾雜點狀色素減退斑及針尖大堅實丘疹,皮損處皮膚輕度萎縮、毛細血管擴張(圖6)。組織學檢查示:表皮角化過度,基底細胞液化變性,真皮多個乳頭內見嗜伊紅物質沉積(圖7)。甲基紫染色:陽性(圖8)。結合臨床診斷:皮膚異色病樣淀粉樣變病。
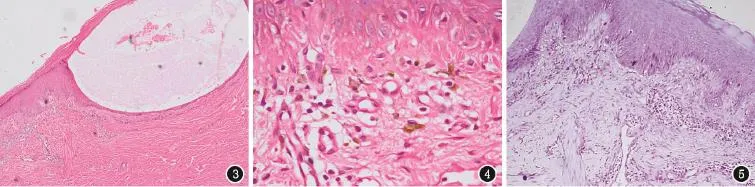
圖3 表皮內皰,皰內見纖維蛋白(HE,×40) 圖4 真皮乳頭見嗜伊紅均質物沉積,散在噬色素細胞,血管周圍少量淋巴組織細胞浸潤(HE,×200) 圖5 甲基紫染色陽性(×40)
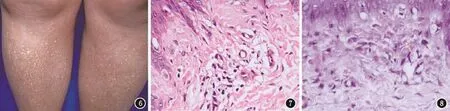
圖6 雙下肢見彌漫性色素沉著斑及苔蘚樣褐色丘疹,夾雜點狀白斑 圖7 基底細胞液化變性,真皮多個乳頭內見嗜伊紅物質沉積(HE,×200) 圖8 甲基紫染色陽性(×200)
討論 皮膚異色病樣淀粉樣變病臨床上有皮膚異色病樣改變,故較為特殊。1936年Marchionini等首先以苔蘚樣皮膚異色病樣淀粉樣變病報道1例,1959年Rockl將其正式命名為皮膚異色病樣淀粉樣變病[1]。Pardo Arranz等認為該病屬常染色體隱性遺傳病,具有家族史的患者發病年齡常較散發病例早,臨床上男性較為多見,且男性患者癥狀常較女性重[2]。本病的發病原因尚未闡明,目前研究表明,遺傳因素、日曬、機體免疫紊亂、環境因素、摩擦因素等均與本病有關[3]。本病的臨床表現為:(1)色素沉著、色素減退斑伴表皮萎縮、毛細血管擴張;(2)發病年齡較早,病程緩慢,好發于四肢;(3)可有苔蘚樣丘疹;(4)可有身材矮小、光敏、水皰及掌跖角化;(5)可伴有瘙癢;(6)真皮乳頭處有淀粉樣物質沉積[4]。
PCA根據患者發病年齡,又分為兩型:一型為出生后至青春期發病,該型除具有上述典型皮損外,尚有身材矮小及光敏;另一型為成年發病[1]。本文兩例患者均具有PCA的典型皮損及組織病理表現,且均為成年發病,故診斷符合皮膚異色病樣淀粉樣變病。本病臨床上不僅需要與原發性皮膚淀粉樣變病的其他亞型相鑒別,也需要與皮損同樣具有皮膚異色病樣改變的疾病(如先天性皮膚異色癥、血管萎縮性皮膚異色病、結締組織病等)相鑒別,和前者的區別在于PCA的皮損有皮膚異色病樣改變,而和后者可通過組織病理及甲基紫染色相區別。有水皰表現者還需要與大皰性類天皰瘡相鑒別,兩者均為張力性水皰,尼氏征陰性,但后者臨床上無皮膚異色病樣改變,組織病理無嗜伊紅物質沉積,免疫熒光陽性。本病無特效治療方法,可避光,口服阿維A酯或阿維A,外用糖皮質激素類制劑或卡泊三醇治療,也可選用調Q激光或脈沖燃料激光,但此法價格高且易復發。李丹曾報道對患者予羥氯喹0.1 g,每天兩次口服;維生素E 50 mg、維生素C 0.1 g,每天三次口服,聯合哈西奈德軟膏外用治療取得一定的效果[5]。本文兩例患者均予維安酯25 mg,每天兩次口服,外用糖皮質激素乳膏及卡泊三醇軟膏治療,兩例患者的丘疹有明顯變平,但色素沉著及減退斑無明顯好轉,目前正隨訪治療中。
[1]趙辨.中國臨床皮膚病學[M].南京:江蘇科學技術出版社,2009.1396.
[2]Pardo Arranz L,Escalonilla García-Patos P,Román Curto C,et al.Familial poikylodermic cutaneous amyloidosis[J]. Eur J Dermatol,2008,18(3):289-291.
[3]吳成,周曉鴻,李文華.皮膚異色病樣淀粉樣變病二例[J].中國皮膚性病學雜志,2012,26(7):641.
[4]Ho MH,Chong LY.Poikiloderma-like cutaneous amyloidosis in an ethnic Chinese girl[J].J Dermatol,1998,25(11):730 -734.
[5]李丹.皮膚異色病樣淀粉樣變病一例[J].臨床皮膚科雜志,2005,34(6):392.
(收稿:2014-11-20 修回:2014-12-20)
Poikiloderma-like cutaneous amyloidosis:two cases report
AI Jing,LI Eryi,WU Tieqiang,GU Youshou,Yang Bin.
Guang Dong Provincial Dermatology Hospital,Guangzhou,510500,China Corresponding author:Yang Bin,E-mail:yangbin101@hotmail.com
One patient was a 49-years-old male and another patient was a 21-years-old female,the course of the disease of whom was 9 years and 3 years respectively.Both patients presented with diffused skin pigmentation speckled with white patches,accompanied with lichenoid papules and vesicles occasionally.There was no similar case in the family.Histopathological examination showed the deposition of amyloid-like substance in the papillary layer.
amyloidosis;poikiloderma
楊斌,E-mail:yangbin101@hotmail.com
廣東省皮膚病醫院,廣東廣州,510500
