適合分解水的釕納米合金催化劑
2016-09-03 03:07:12李海生李冠亞李立本
中國科技信息 2016年10期
李 永 李海生 李冠亞 李立本
?
適合分解水的釕納米合金催化劑
李 永 李海生 李冠亞 李立本
本文基于密度泛函理論,用第一性原理計算得到了H2O-XRun-1(n=2-14)團簇(X= Rh、Pd、Au)的最穩定幾何結構,并計算了水分子在Ru合金團簇上吸附能。研究結果表明:水分子在具有偶數個原子的合金團簇上具有最大的吸附能,它們分別是H2O- RhRu5、H2O-PdRu3以及 H2O-AuRu7體系。結合前期研究發現:水分子在RhRu5、PdRu3以及AuRu7合金團簇上的吸附能遠大于水分子在Ru塊體表面的吸附能,大于水分子在純Run(n=2-14) 上的吸附能,而略小于H2O分子在PtRu7團簇的吸附能。本文研究表明合金效應加強了水分子的吸附作用,以保證水分子被分解之前不會以分子形式脫離吸附,這些Ru合金團簇有望成為適合分解水的高效催化劑。
由于城市霧霾嚴重和全球能源短缺,催化分解水產氫是目前國內外能源領域研究的熱點之一,利用TiO2光催化分解水產氫,是其中的一個重要方向。在理論上,利用第一性原理研究TiO2光催化分解水是一種非常有效的方法。在TiO2上沉積過渡金屬顆粒,不僅可以降低TiO2的帶隙,也可以降低電子-空穴符合的幾率,是尋找分解水的高效催化劑的一種有效方法。在研究與水分子作用較強的金屬催化劑方面,以前的工作有大量介紹。Feibelman 研究表明吸附在Ru(0001)表面的水是半分解的,其中一個OH 鍵斷開,剩余孤立的H 原子直接與金屬表面結合,這是催化產生氫能源的關鍵一步。Meng等人詳細研究了與水分子作用較強的一些過渡金屬,發現,水分子與Ru、Rh、Pd、Pt 和Au 表面的結合能逐漸減弱,但是水分子在這些過渡金屬表面幾乎不分解。Ranjit 等人通過實驗發現光催化合成氨氣的產率受在TiO2上沉積的金屬本身屬性影響,氨氣產率取決于中間產物中金屬與氫原子成鍵強弱,金屬與氫原子成鍵越強,氨氣產率越高。這個實驗啟示我們先找到與水分子結合較強的合金團簇,然后直接將其沉積在TiO2襯底上,即可得分解水分子的高效催化劑。與相對應的過渡金屬塊體材料相比,團簇的電子結構及穩定性等的物理及化學性質存在很大差異。過渡金屬團簇表面積比較大,在催化反應式活性位更多。例如,用金屬團簇替代其塊體表面催化分解水產氫的效果會更好。
在合金催化領域,根據Schmid的理論,不同配位數原子的相互作用可以產生與一元金屬不同的特性,即協同效應。據Nerlov等人報道,二元合金表面的催化活性比較高。
另據Zhang等人的研究,合金比純的單一金屬催化活性高。也就是說,提高催化活性的另一種有效方法是合金效應。Desai 等人利用第一性原理研究發現水在Pt-Ru合金塊體表面的分解勢壘比水在Pt塊體表面或者Ru塊體表面上的分解勢壘降低很多。另一方面,在催化分解水時,水分子在合金團簇上較大的吸附能,可以保證水分子的OH鍵在斷裂前不容易以分子形式從合金團簇脫附,從而也比較容易達到催化分解水的目的。本文基于之前的研究成果 (我們之前計算H2O-PtRun(n=1-13)團簇和H2O-Run(n=1-14)團簇的吸附能,其中H2O分子在Ru8和PtRu7團簇上的吸附能最大,其值為1.06 eV(另文發表))。 本文又深入研究了H2O-XRun-1(n=2-14) 團簇(X= Rh、Pd和Au) 的吸附性能。本文只研究了前一部分,即水分子在這些過渡金屬合金團簇上的吸附性能。本文的研究結果有望為實驗上尋找分解水的高效合金催化劑奠定了理論基礎。
計算方法
采用基于密度泛函理論的第一性原理計算方法,使用VASP程序軟件包對H2O-XRun-1(n=2-14)(包括合金)團簇(X=Pd、Rh、Au)進行總能量計算。從離子受力情況出發,計算出離子在新平衡位置總能的最小值,以優化合金團簇結構。用GGA-PBE方法來描述電子的交換關聯勢。用Vanderbilt超軟贗勢描述離子實,波函數由平面波展開。電子結構弛豫的能量收斂標準和每個離子上力的收斂標準分別為10-4eV和0.02 eV/?,所有計算采用自旋極化。通過檢測能量能否收斂,檢測上述設定能否保證計算精度。吸附能Eads的大小根據公式Eads=-[E(H2OXRun-1)-E(H2O)-E(XRun-1)]計算得到,其中E(H2O/XRun-1)為H2O-XRun-1團簇的總能量,E(H2O)和E(XRun-1)分別為水分子的能量和XRun-1團簇的總能量。
結果與討論
H2O-RhRun-1(n=2-14)的幾何結構及吸附能
圖1為RhRun-1(n=2-14)的穩定幾何結構和吸附能。由圖1可知,隨著Ru原子數的增加,H2ORhRun-1(n=2-14)團簇的穩定幾何結構由平面結構逐漸傾向于簡單立方體結構轉變,吸附能有先增大后減小的趨勢。當n=6,Ru原子數為5時,H2O-RhRun-1(n=2-14)團簇的吸附能達到了極大值,其數值為0.979 eV。從圖中還可看出,H2O-RhRu5團簇為三棱柱結構,可作為分解水高效合金催化劑的候選材料。
H2O-PdRun-1(n=2-14)的幾何結構及吸附能
圖2為PdRun-1(n=2-14)的穩定幾何結構和吸附能。從圖2中可以看出,隨著Ru原子數的增加,H2O-PdRun-1(n=2-14)團簇的穩定幾何結構由平面結構逐漸向簡單立方體結構轉變,吸附能有先增大后減小的趨勢。當n=4,即Ru原子數為3時,H2OPdRun-1(n=2-14)團簇的吸附能達到了極大值,其數值為0.814 eV。也就是說,水分子與PdRu3團簇吸附能是H2O-PdRun-1團簇最大的,PdRu3可作為分解水高效合金催化劑的候選材料。
H2O-AuRun-1(n=2-14)的幾何結構及吸附能
圖3為AuRun-1(n=2-14)的穩定幾何結構和吸附能。從圖3可以看出,隨著Ru原子數的增加,H2O-AuRun-1(n=2-14)團簇的穩定幾何結構由平面結構逐漸傾向于簡單立方體結構轉變。當n=8,即Ru原子數為7時,H2O-AuRun-1(n=2-14)團簇的吸附能達到了極大值,其數值為0.931 eV。也就是說,水分子與AuRu7團簇吸附能是H2O-AuRun-1團簇最大的,AuRu7也可作為分解水的高效合金催化劑。
H2O在XRun-1(n=2-14)團簇的吸附能比較
圖4為 Run、RhRun-1、PdRun-1、PtRun-1、AuRun-1(n=2-14)與H2O分子之間的結合能隨著n的變化關系圖。我們之前研究了純Run團簇、PtRun-1(n=2-14)團簇與水分子的吸附能。在此,本文將這些結果作比較,以求找到作為分解水的高效合金催化劑。
由圖4可以看出,水分子在RhRu5、PdRu3以及AuRu7合金團簇上的吸附能大于水分子在純Run(n=2-14) 上的吸附能,而略小于H2O分子在PtRu7團簇的吸附能(1.06 eV),但是這些吸附能大于文獻中報道的水在純Ru團簇上的分解勢壘,并且遠大于H2O分子在金屬塊體上的吸附能。因此這些合金團簇RhRu5、PdRu3、AuRu7以及PtRu7可作為分解水的高效合金催化劑。
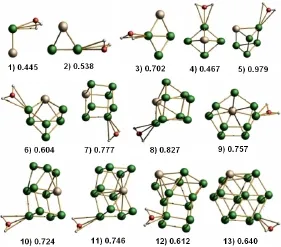
圖1 H2O-RhRun團簇的吸附能和特征鍵長(綠、灰、紅、小灰白色代表Ru、Rh、O和H原子)
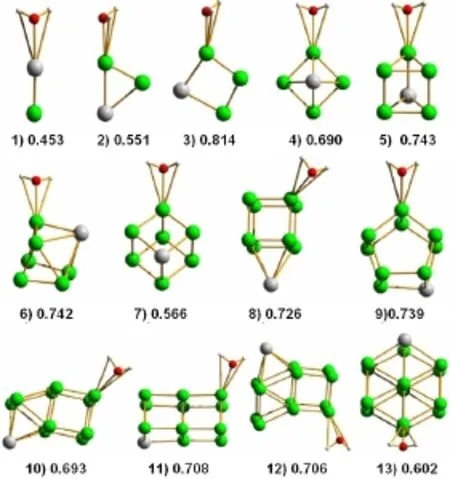
圖2 H2O-PdRun團簇吸附能(綠、灰、紅、小灰白色代表Ru、Pd、O和H原子)

圖3 H2O-AuRun團簇結合能和特征健長,綠、黃、紅、灰白分別是釕、金、氧、氫原子

圖4 Run、RhRun-1、PdRun-1、PtRun-1、AuRun-1與H2O分子之間的吸附能隨著n的變化
結語
本文采用第一性原理研究了H2O-XRun-1(n=2-14)團簇(其中X為Pd、Rh、Au原子)的穩定幾何結構和吸附能。結果表明:水分子喜歡吸附在這些合金團簇上具有低配位數的釕原子上。水分子在RhRu5、PdRu3以及AuRu7合金團簇上的吸附能在各自合金體系中是最大的,大于水分子在純Run(n=2-14) 上的吸附能,而略小于H2O分子在PtRu7團簇的吸附能(1.06 eV),但是這些吸附能大于文獻中報道的水在純Ru團簇上的分解勢壘,并且遠大于H2O分子在金屬塊體上的吸附能。這樣可以保證水分子在分解前不會以分子形式脫離表面吸附,因此這些合金團簇RhRu5、PdRu3、AuRu7以及PtRu7可作為分解水的高效合金催化劑。本文的研究結果可以為實驗上高效合金催化劑來催化分解水提供理論支持。

李 永 李海生 李冠亞 李立本
河南科技大學物理工程學院
10.3969/j.issn.1001-8972.2016.10.002
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
哲學評論(2021年2期)2021-08-22 01:53:34
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
中華詩詞(2019年7期)2019-11-25 01:43:04
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
