UPLC-Q-TOF-MS法分析芩百清肺濃縮丸中的化學成分
2016-09-06 05:22:04褚衍濤魏文峰霍金海王偉明黑龍江省中醫藥科學院黑龍江哈爾濱150036
中成藥 2016年6期
褚衍濤, 魏文峰, 霍金海, 王偉明(黑龍江省中醫藥科學院,黑龍江哈爾濱150036)
?
UPLC-Q-TOF-MS法分析芩百清肺濃縮丸中的化學成分
褚衍濤, 魏文峰, 霍金海, 王偉明*
(黑龍江省中醫藥科學院,黑龍江哈爾濱150036)
目的 建立超高效液相色譜-質譜聯用(UPLC-Q-TOF-MS)法分析芩百清肺濃縮丸(黃芩、百部、紫菀等)中的化學成分。方法 分析采用Acquity UPLC BEH C18色譜柱(2.1 mm×100 mm,1.7μm);流動相為0.1%甲酸水(A)-0.1%甲酸乙腈 (B),梯度洗脫;體積流量0.3 mL/min;柱溫35℃。結果 共鑒定出78個化合物,14個來自黃芩,29個來自百部,10個來自麥冬,13個來自紫菀,3個來自桔梗,9個來自地龍。結論 該方法準確快速,可用于芩百清肺濃縮丸的鑒定分析。
芩百清肺濃縮丸;化學成分;UPLC-Q-TOF-MS
芩百清肺濃縮丸是黑龍江省中醫藥科學院自主研發,國家食品藥品監督管理局批準的第一個用于臨床治療小兒支原體肺炎的中藥新藥,由黃芩、百部、紫菀、桔梗、麥冬、地龍6味中藥組成,具有清熱解毒、降氣平喘、潤肺止咳之功效。前期實驗表明,其能提高免疫功能和促進氣道上皮細胞的修復,能有效地減輕支原體對肺部組織的炎性病變,促進上皮細胞的恢復[1-3]。但是,芩百清肺濃縮丸的化學成分不明確,制約了藥效物質基礎及作用機制研究。
近年來,色譜-質譜聯用技術發展迅速,日益成為天然藥物和中藥方劑中化學成分快速分離和鑒定的有力手段[4-5]。本實驗應用超高效液相色譜-質譜聯用(UPLC-Q-TOF-MS)法首次對芩百清肺濃縮丸進行研究,根據精確分子量、同位素豐度比、分子碎片峰,參考相關文獻、對照品色譜保留時間及二級譜圖,共鑒定了78個化合物,為該藥物的藥效物質基礎及作用機制研究奠定了基礎。
1 儀器和試藥
Waters ACQUITYTMUPLC色譜儀(美國Waters公司);AB SCEIX Trip1e-TOFTM 5600+高分辨質譜儀,配有ESI源和APCI源(美國AB SCIEX公司);BP211D型精密分析天平 (賽多利斯科學儀器北京有限公司);KQ-800KDB型超聲波清洗儀(昆山市超聲儀器有限公司)。
甲醇、乙腈為色譜純 (德國Merck公司);甲酸為色譜純(美國Fisher公司);水為蒸餾水(廣州屈臣氏食品飲料有限公司)。
黃芩苷 (批號715-200111)、槲皮素 (批號100081-200406)、黃芩素 (批號111595-200402)、漢黃芩素 (批號111514-200403)、咖啡酸 (批號110885-200102)、綠原酸 (批號110753-200212)、山奈酚 (批號110861-200406)、次黃嘌呤 (批號140661-200903)對照品 (中國食品藥品檢定研究院)。金剛大堿對照品 (自制,含有量大于98%)。
芩百清肺濃縮丸 (批號20150401,3 g/袋,黑龍江省中醫藥科學院制劑室)。
2 實驗方法
2.1色譜條件 Waters ACQUITY UPLCBEH C18色譜柱(2.1 mm×100 mm,1.7μm),VanGuard Pre-Co1umn預柱(2.1 mm×5 mm,1.7μm);流動相0.1%甲酸水 (A)-0.1%甲酸乙腈(B),梯度洗脫(0→4 min,95%→73%A;4→5 min,73%A;5→10 min,73%→50%A;10→13 min,50%→30%A;13→14 min,30%A;14→20 min,30%→0%A);柱溫35℃;體積流量0.3 mL/min;進樣量3μL。
2.2質譜條件 電噴霧正、負離子模式,離子源電壓分別為5 500、4 500 V,離子源溫度550℃;霧化氣體N2,霧化氣、輔助氣壓力379.17 kPa,氣簾氣壓力241.29 kPa;裂解電壓±80 V;碰撞能量±35 eV,碰撞能量擴展15 eV。TOF MS掃描范圍100~1 500 Da,IDA設置響應值超過100 cps的8個最高峰進行二級質譜掃描;Product Ion掃描范圍50~1 500 Da,開啟動態背景扣除。數據采集軟件為Ana1yst TF 1.6 software,數據處理軟件為Peakview 2.0/masterview1.0 software。
2.3全方供試品溶液制備 精密稱取芩百清肺濃縮丸粉末1 g(過40目篩),加入30 mL75%甲醇,超聲30 min,13 000 r/min離心10 min,取上清液,即得。
2.4單味藥供試品溶液的制備 稱取各單味藥,按制備工藝制成各單味藥的凍干粉,根據處方比例稱取,按 “2.3”項下方法處理,取上清液,即得。
2.5數據分析策略 (1)依據UPLC-Q-TOF-MS得到色譜峰的同位素豐度和一級、二級質譜,采用Formu1a Finder計算其精確的分子質量,推測其可能的化學組成,誤差控制在百萬分之5以內;(2)根據國內外相關文獻,收集黃酮、生物堿、皂苷及其他來源于組方6味中藥中的化學成分信息,建立化學結構數據庫,包括英文名稱、分子式、精確分子量、植物來源、CAS號及所屬類型。通過與文獻中的信息對照,快速指認可能的化學成分;(3)通過與對照品保留時間和二級質譜數據對比,確認推斷結果。對于一些無法獲得對照品的化合物,依據MS/MS碎片信息以及反相色譜行為,進一步推斷其結構。
3 結果
3.1UPLC-QTOF-MS成分推斷 正負離子掃描模式下的總離子流圖見圖1。

圖1 總離子流圖Fig.1 Total ion current chromatogram s
3.2芩百清肺濃縮丸成分分析 共鑒定出78個化合物,其中14個來自黃芩,29個來自百部,10個來自麥冬,13個來自紫菀,3個來自桔梗,9個來自地龍。結果見表1。
3.3主要色譜峰的質譜分析
3.3.1黃酮類 該類化合物主要來自黃芩和麥冬,大多為含有葡萄糖酸和葡萄糖的黃酮苷。黃芩中黃酮裂解的文獻報道較多[6-8],可見丟失相應葡萄糖酸或葡萄糖殘基的苷元離子,其裂解規律與相應苷元的準分子離子裂解一致,中間環的裂解是黃酮的典型裂解方式。另外,多數苷元均觀察到脫水或脫甲基離子,因為環的裂解,可見到脫 CO、脫C2H2O離子。以黃芩苷為例闡述裂解過程,正離子模式下給出的準分子離子峰m/z 447.089 2[M+ H]+,特征碎片m/z 271[M+H-176]+,表明該準分子離子丟失一個葡萄糖醛酸的中性碎片。接著發生中性丟失,脫掉H2O,產生碎片離子m/z253[M+H-C6H8O6-H2O]+;苷元C環裂解,脫掉C2、C3及B環產生碎片離子m/z169[M+H-C6H8O6-C8H6]+,與文獻 [8]一致,推測其為黃芩苷。進一步與黃芩苷標品數據對比,其色譜、質譜行為一致,故鑒定此峰為黃芩苷,見圖2~3。
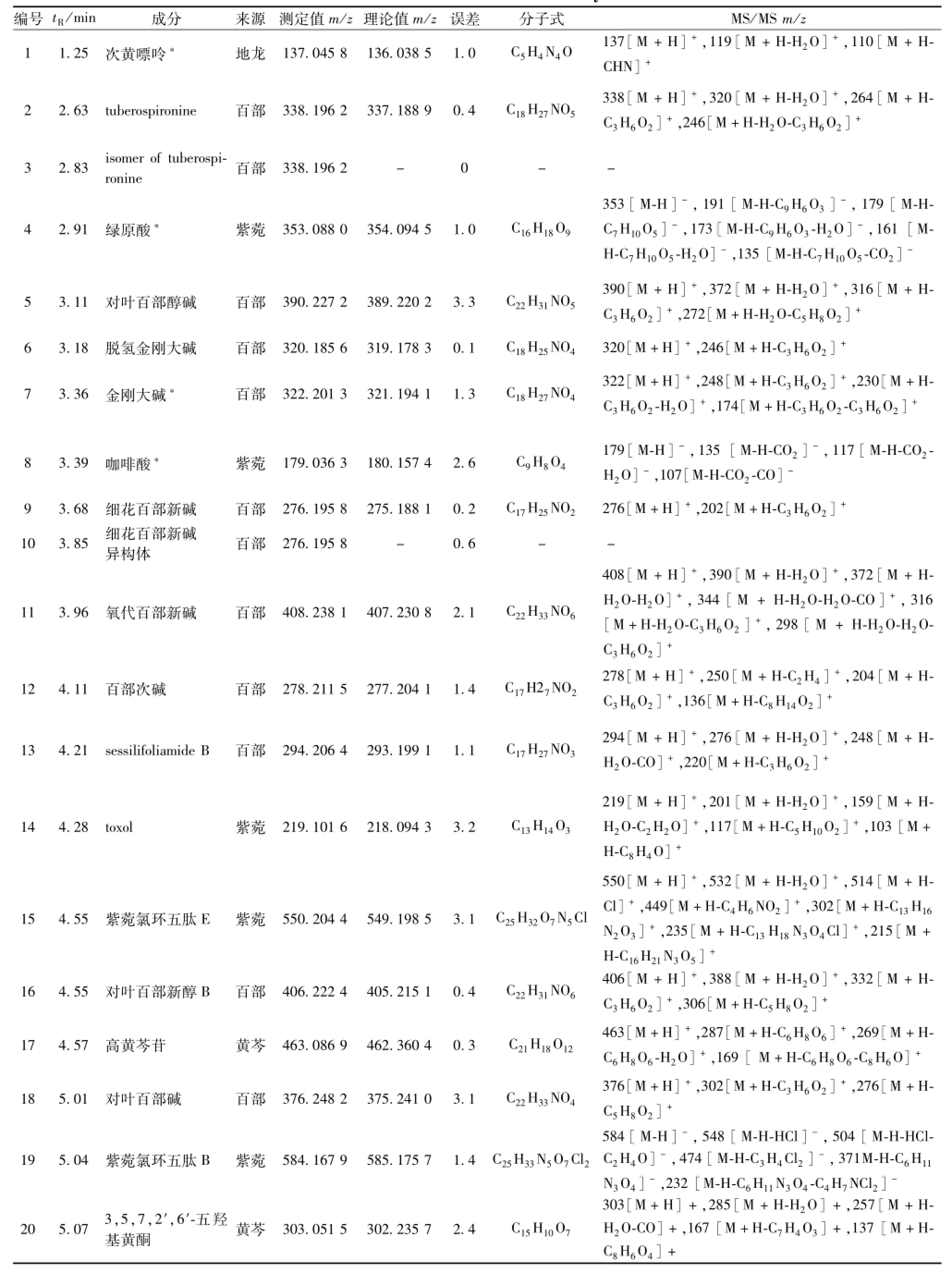
表1 成分分析結果Tab.1 Resu lts of constituent analysis

續表1
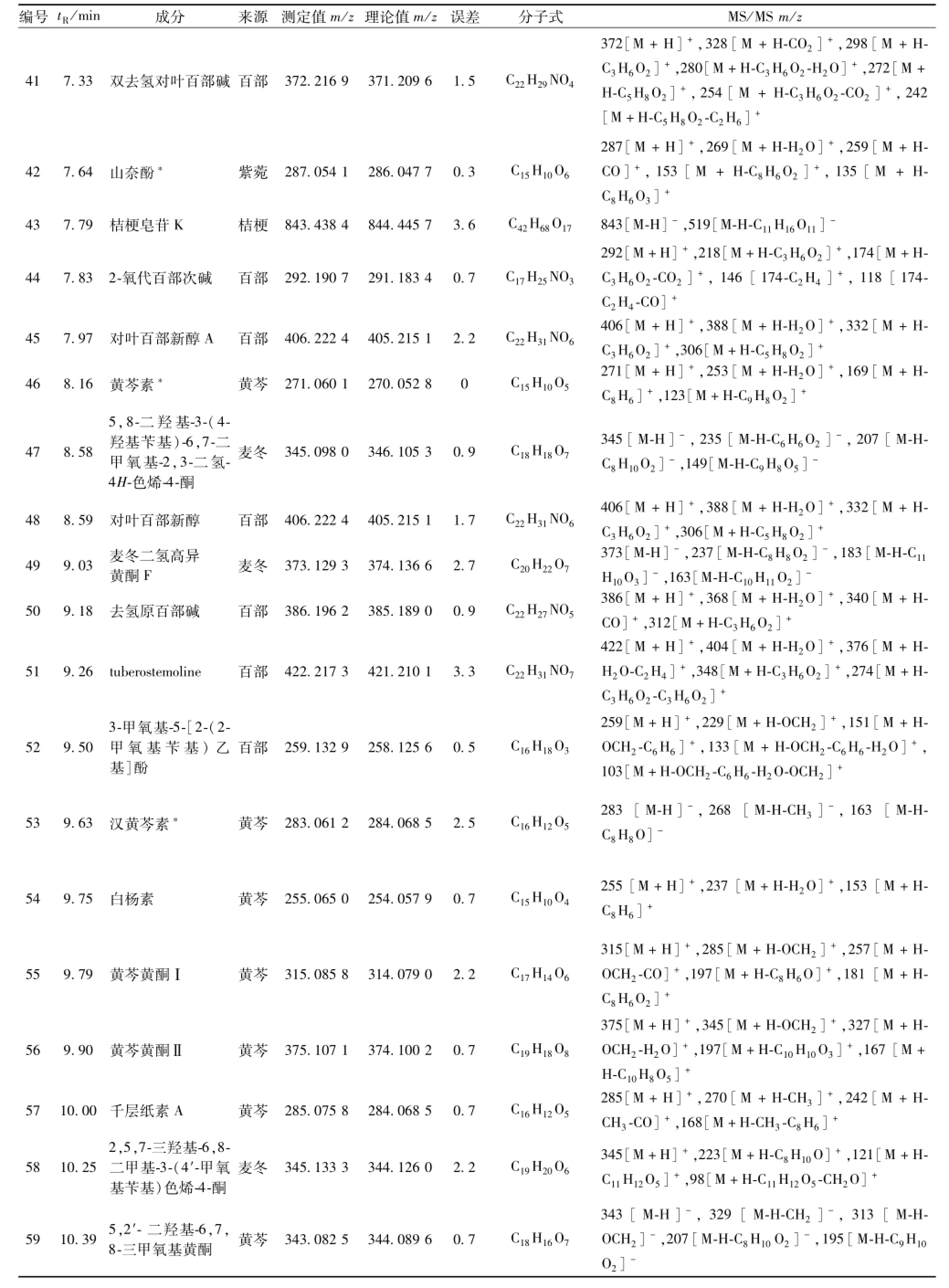
續表1
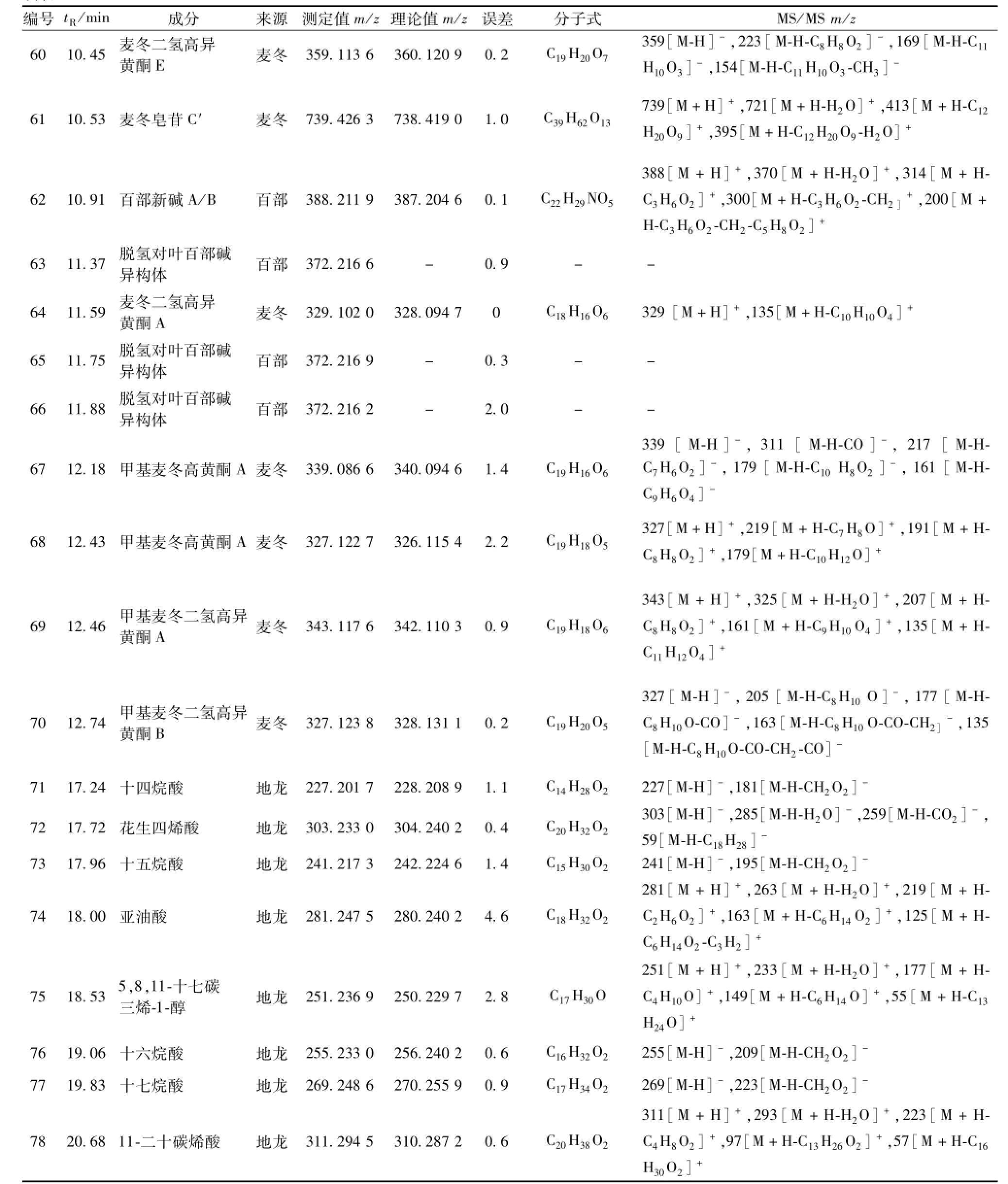
續表1

圖2 黃芩苷正離子模式下的ESI-MS2質譜圖Fig.2 ESI-MS2mass spectrum of baicalin in a positivem ode
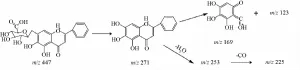
圖3 黃芩苷的裂解途徑Fig.3 Fragmentation pathway of baicalin
麥冬中高異黃酮是較特殊的一類,其母體結構僅比異黃酮多一個碳原子,但其裂解方式與黃酮有明顯區別,而且其與二氫高異黃酮也有各自獨特的裂解途徑。結合文獻 [9-11],共推測出9個高異黃酮類化合物。結合文獻 [9],對于二氫高異黃酮,其首先斷裂C3-9鍵,丟失B環,接著發生中性丟失脫掉CO;對于C2與C3間為雙鍵的高異黃酮,其主要裂解方式為首先發生中性丟失脫掉CO,接著再發生C3-9或者C9-1'斷裂丟失B環。以甲基麥冬黃酮B為例,見圖4~5。

圖4 甲基麥冬黃酮B負離子模式下的ESI-M S2質譜圖Fig.4 ESI-MS2mass spectrum of methylophiopogonanone B in a negativemode
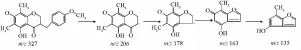
圖5 甲基麥冬黃酮B的裂解途徑Fig.5 Fragm entation pathway of methylophiopogonanone B
3.3.2生物堿類 該類化合物主要來自百部,是百部屬植物的主要化學成分及活性成分,大多具有吡咯 [1,2-α]氮雜環或吡啶 [1,2-α]氮雜環的母核,其質譜裂解行為報道較少[12]。該類化合物二級質譜中均可見 [M+H-100]+峰,由準分子離子脫去一個α-甲基-γ-內酯環而來;還可見[M+H-74]+峰,由準分子離子經氫原子轉移引發四元環重排,脫去CH3CH=C(OH)2產生;還可以看到脫甲基、脫水、脫CO等碎片離子峰。以金剛大堿為例,其正離子模式下給出 [M+H]+為m/z322,特征碎片m/z248為氫原子轉移后發生四元重排丟失m/z 74的碎片產生的,進一步相繼丟失H2O和CO2后生成m/z230和m/z204的碎片離子,經異構后再次丟失m/z74的碎片,產生m/z 174[M+H-74-74]+峰。結合文獻 [13],初步判定為金剛大堿,進一步與標準品對比,色譜、質譜行為一致,故鑒定為金剛大堿,見圖6~7。

圖6 金剛大堿正離子模式下的ESI-MS2質譜圖Fig.6 ESI-MS2mass spectrum of croom ine in a positivemode
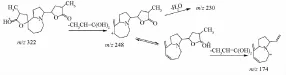
圖7 金剛大堿的裂解途徑Fig.7 Fragmentation pathway of croom ine
3.3.3其他類 地龍中大多為飽和與不飽和脂肪酸,主要以脫羧裂解為特征。紫菀中主要為萜類、五肽及酚酸類,其中對五肽類化合物的報道大多為其化學成分及藥理作用,未見裂解規律的描述[14];酚酸類成分報道較多,其裂解規律比較明確,結合文獻 [15],鑒定為綠原酸、咖啡酸等酚酸類成分。
4 討論
本實驗推測,所鑒定的化合物均可在單味藥中得到歸屬,實驗過程中亦發現在全方的基峰(BPC)圖上可以檢測到單味藥中沒有的新成分,但由于其豐度值較低,難以獲得二級譜圖等信息,故不能確定結構。桔梗中的主要成分為桔梗皂苷類,其結構不穩定,易發生源內裂解,測定時需要的DP值及CE值較低。考慮到其他大部分化合物的測定要求,去簇電壓 (DP)設為±80 V,碰撞能量(CE)設為±35 eV,碰撞能量擴展(CES)設為15 eV,推測可能是桔梗類成分檢測不到的重要因素,尚需開展深入研究。
結合文獻分析,黃芩苷具有直接誘殺支原體的作用,其對支原體的MIC為0.97~3.90 g/L[16],而且黃芩素和黃芩苷均具有解熱、鎮痛、抗炎等作用[17]。麥冬中的高異黃酮類具有明顯的抗炎等藥理活性[18],而皂苷類化學成分在抗炎和免疫調節等多方面均能發揮作用[19]。百部生物堿類可以抑制咳嗽反射而具有鎮咳的作用[20]。紫菀中萜類等成分是化痰止咳的主要活性成分[21]。地龍中的次黃嘌呤是平喘的主要活性成分,而高度不飽和脂肪酸等具有調節免疫系統,增強免疫功能[22],因此,這些化合物可能是芩百清肺濃縮丸的主要藥效物質基礎。
實驗過程中發現,部分成分互為同分異構體,如63、65、66號化合物,無法確定其具體結構。但部分有相關文獻報道,如16、45、48號化合物[13],依據其在反相色譜上的行為,16號首先流出色譜柱,其后則是45號,48號最后流出,由此推測同分異構體的歸屬。
本實驗應用UPLC-Q-TOF-MS聯用技術對芩百清肺濃縮丸中的化學成分進行初步定性分析,通過與部分對照品數據比較及文獻分析,共鑒定或推斷了78個化合物的結構。結果表明,該方法簡便、準確,可為芩百清肺濃縮丸的藥效物質基礎研究奠定基礎。
[1]蒙艷麗,王 欣,王偉明.芩百清肺濃縮丸對支原體作用[J].中成藥,2014,36(1):36-39.
[2]李繼昌,董 龍,楊 陽,等.芩百清肺濃縮丸對支原體肺炎模型鼠肺組織影響的研究[J].中國預防獸醫學報,2004,26(3):222-225.
[3]王偉明,張洪娟,唐慧蘭.芩百清肺濃縮丸主要藥效學研究[J].黑龍江中醫藥,2003(6):42-43.
[4]Cheng H Y,Chen JH,Yang H H,etal.Identification ofactive components in Styela clava by HPLC-ESI-TOF/MS and the study on their HPLC specific chromatograms[J].Acta Pharm Sin,2010,45(10):1285-1289.
[5]康利平,趙 陽,余河水,等.采用UPLC-Q-TOF/MSE鑒別芪藶強心膠囊有效部位的化學成分[J].藥學學報,2011,46(10):1231-1236.
[6]Es-Safi N E,Kerhoas L,Einhorn J,et al.App1ication of ESI/MS,CID/MS and tandem MS/MS to the fragmentation study oferiodictyo17-O-g1ucosy1-(1→2)-g1ucoside and 1uteo-1in 7-O-g1ueosy1-(1→2)-g1ucosid[J].Int J Mass Spec,2005,247(1-3):93-100.
[7]劉國柱.中藥中黃酮類化合物定性分析策略[D].長沙:湖南師范大學,2010.
[8]沈 紅,段金廒,錢大偉,等.黃芩及復方野馬追膠囊中黃酮類成分的LC-MS/MS分析[J].藥物分析雜志,2009,29(9):1425-1429.
[9]張小燕.中藥麥冬的活性成分分析[D].南京:南京中醫藥大學,2004.
[10]Ye M,Guo D,Ye G,et al.Ana1ysis of homoisof1avonoids in Ophiopogon japonicus by HPLC-DAD-ESI-MSn[J].J Am Soc Mass Apectrom,2005,16(2):234-243.
[11]Zhou Y F,Qi J,Zhu D N,et al.Homoisof1avonoids from Ophiopogon japonicu s and its oxygen free radica1s(OFRs)scavenging effects[J].Chin J Nat Med,2008,6(3):201-204.
[12]彭蜀瑩,沈建華,葉 陽.4種百部生物堿的質譜行為研究[J].分析化學,2006,34(4):497-502.
[13]鐘 瑩.對葉百部化學成分及質量研究[D].廣州:廣州中醫藥大學,2010.
[14]庫爾班江,歐陽艷,努爾買買提.紫菀屬植物化學成分及藥理作用研究進展[J].中國野生植物資源,2010,29(2):1-4.
[15]王祝偉.紅毛五加化學成分及其多維指紋圖譜研究[D].沈陽:沈陽藥科大學,2005.
[16]孫艷平,董秀華.黃芩苷體外誘導肺炎支原體耐藥的實驗研究[J].中醫藥信息,2010,27(4):115-116.
[17]辛文妤,宋俊科,何國榮,等.黃芩素和黃芩苷的藥理作用及機制研究進展[J].中國新藥雜志,2013,22(6):647-653.
[18]姜洪波,黃 靜,郭明娟,等.天然高異黃酮的研究進展[J].藥學學報,2007,42(2):118-126.
[19]于學康.麥冬的藥理作用研究進展[J].天津藥學,2012,24(4):69-70.
[20]Lin L G,Yang X Z,Tang C P,et al.Antibacteria1 sti1benoids from the roots of Stemona tuberose[J].Phytochemistry,2008,69(2):457-463.
[21]彭文靜,辛蕊華,任麗花,等.紫菀化學成分及藥理作用研究進展[J].動物醫學進展,2015,36(3):102-107.
[22]祝未名.中藥地龍的活性成分與藥理作用研究[J].海峽藥學,2013,25(4):25-26.
Analysis of chem ical constituents in Qinbai Qingfei Concentrated Pills by UPLCQ-TOF-MS
CHU Yan-tao, WEIWen-feng, HUO Jin-hai, WANGWei-ming*
(Heilongjiang Provincial Academy of Chinese Medical Sciences,Harbin 150036,China)
AIM To estab1ish an u1tra performance 1iquid chromatography quadrupo1e time of f1ightmass spectrometry(UPLC-Q-TOF-MS)method for ana1yzing the chemica1 constituents in Qinbai Qingfei Concentrated Pi11s(Scutellariae Radix,Stemonae Radix,Asteris Radix et Rhizoma,etc.).M ETHODS The ana1ysiswas performed on an Acquity UPLC BEH C18co1umn(2.1 mm×100mm,1.7μm),mobi1e phase was0.1%formic acid aqueous so1ution(A)-0.1%formic acid and acetonitri1e(B)with gradiente1ution,f1ow rate was 0.3 mL/min,and co1umn temperature wasmaintained at 35℃.RESULTS Tota1seventy-eight constituents were identified,fourteen of which came from Scutellariae Radix,twenty-nine of which came from Stemonae Radix,ten of which came from Ophiopogonis Radix,thirteen ofwhich came from AsterisRadix et Rhizoma,three ofwhich came from Platycodonis Radix,nine of which came from Pheretima.CONCLUSION Thismethod is accurate and rapid,which can be used for the identification and ana1ysis of QinbaiQingfei Concentrated Pi11s.
Qinbai Qingfei Concentrated Pi11s;chemica1 consituents;UPLC-Q-TOF-MS
R284.1
A
1001-1528(2016)06-1303-08
10.3969/j.issn.1001-1528.2016.06.021
2015-11-27
國家 “重大新藥創制”科技重大專項 (2010ZX09101-104);黑龍江省青年基金 (QC2011C114)
褚衍濤 (1990—),男,碩士生,從事中藥新藥研發工作。Te1:(0451)55653086-6902,E-mai1:chuyantao2010@126.com
王偉明 (1966—),女,博士,研究員,從事中藥新藥研發工作。Te1:(0451)55665478,E-mai1:zyyjy@163.com
