精氨酸缺陷型菌株發(fā)酵生產(chǎn)反式-4-羥脯氨酸
2017-02-15 05:37:04王曉姣張震宇孫付保于林
食品與發(fā)酵工業(yè) 2017年1期
王曉姣,張震宇,孫付保,于林
(江南大學(xué) 生物工程學(xué)院,江蘇 無錫,214122)
精氨酸缺陷型菌株發(fā)酵生產(chǎn)反式-4-羥脯氨酸
王曉姣,張震宇*,孫付保*,于林
(江南大學(xué) 生物工程學(xué)院,江蘇 無錫,214122)
為了獲得高產(chǎn)反式-4-羥脯氨酸的菌株,基于大腸桿菌的代謝網(wǎng)絡(luò)模型的指導(dǎo),以大腸桿菌E.coliBL21(DE3)ΔputA為出發(fā)菌株,通過基因敲除技術(shù)成功敲除argB基因,阻斷L-脯氨酸合成的前體物L(fēng)-谷氨酸的分支代謝途徑,增加L-脯氨酸合成的代謝流,構(gòu)建了精氨酸缺陷型菌株E.coliBL21(DE3)ΔputAΔargB。同時(shí)轉(zhuǎn)入表達(dá)質(zhì)粒pUC19-proB2A-Ptrp2-hyp,該質(zhì)粒含有突變基因proB2,該突變基因所編碼的谷氨酸激酶受L-脯氨酸的反饋抑制作用顯著降低。搖瓶發(fā)酵結(jié)果表明,在外源添加600 mg/LL-精氨酸時(shí),該重組菌株產(chǎn)反式-4-羥脯氨酸的量達(dá)到312.67 mg/L,較菌株E.coliBL21(DE3)ΔputA/pUC19-proB2A-Ptrp2-hyp提高了25.29%。
大腸桿菌1;反式-4-羥脯氨酸2;基因敲除3;脯氨酸4
反式-4-羥脯氨酸(trans-4-hydroxyproline,Hyp)為亞氨基酸,是L-脯氨酸經(jīng)反式-4-羥化酶(proline-4-hydroxylase,hyp) 羥基化的產(chǎn)物,歸屬高附加值小品種氨基酸類[1]。Hyp在食品營養(yǎng)、化工生產(chǎn)、護(hù)膚美容業(yè)以及醫(yī)藥保健具有廣泛的應(yīng)用[2-3]。由于其潛在的醫(yī)藥、食品價(jià)值以及市場(chǎng)需求,促進(jìn)了反式-4-羥脯氨酸的研究步伐。
脯氨酸作為羥脯氨酸合成的底物,前期實(shí)驗(yàn)室生產(chǎn)合成羥脯氨酸都需要外源添加脯氨酸[4-5]。脯氨酸的添加不僅導(dǎo)致生物法合成羥脯氨酸的成本大大提高,而且外源添加的脯氨酸大部分并不能被微生物所利用,造成了資源的浪費(fèi)。
基于對(duì)代謝調(diào)節(jié)的認(rèn)識(shí),可以在基因水平和酶學(xué)水平上對(duì)Hyp的合成途徑進(jìn)行理性操作。目的產(chǎn)物的積累一般有2種方式,積累關(guān)鍵前體和降低副產(chǎn)物生成:L-谷氨酸是L-脯氨酸生物合成的前體物質(zhì),同時(shí)也是L-精氨酸、L-鳥氨酸和L-瓜氨酸生物合成的前體物質(zhì),其胞內(nèi)濃度決定了L-脯氨酸的合成速率[6]。因此,削弱競(jìng)爭(zhēng)代謝途徑,阻斷L-谷氨酸到L-精氨酸的合成代謝流,使得L-谷氨酸到L-脯氨酸合成分支代謝流量增強(qiáng),使得碳源、氮源等更多的用于脯氨酸的合成,可以提高L-脯氨酸的產(chǎn)量[7]。谷氨酸是脯氨酸和精氨酸的共同前體物(圖1)。為提高脯氨酸的產(chǎn)量可以采用基因敲除等方法切斷谷氨酸合成精氨酸的途徑,從而為脯氨酸的生產(chǎn)提供更多的原料。N-乙酰谷氨酸激酶(N-Acetylglutamate kinase EC 7 2.8,NAGK),是精氨酸合成途徑的第2個(gè)酶。該酶由argB基因編碼,是精氨酸途徑中的關(guān)鍵酶。該酶不僅被精氨酸反饋抑制而且也被其反饋?zhàn)瓒鬧8-9],而且是精氨酸生產(chǎn)過程中被精氨酸抑制的唯一限速酶。本文選擇敲除精氨酸代謝途徑中的argB基因,切斷精氨酸代謝途徑,使得脯氨酸大量積累,從而獲得羥脯氨酸高產(chǎn)菌株。

圖1 大腸桿菌中反式-4-羥脯氨酸生物合成途徑Fig.1 The biosynthetic pathways of Hyp in E. coli
1 材料與方法
1.1 材料
1.1.1 菌株與質(zhì)粒
本實(shí)驗(yàn)用到的菌株[5]及質(zhì)粒[10]見表1,其中E.coliBL21(DE3)作為宿主,質(zhì)粒pUC19作為基因表達(dá)載體。

表1 實(shí)驗(yàn)所用菌株和質(zhì)粒
1.1.2 試劑及儀器
葡萄糖、NaCl、MgSO4、K2HPO4、(NH4)2SO4、瓊脂粉、CaCl2、FeSO4、正丙醇、氯胺T、高氯酸、對(duì)二甲氨基苯甲醛、異丙醇、NH4Cl等購自上海國藥。胰蛋白胨、酵母抽提物、瓊脂糖、氨芐青霉素鈉、硫酸卡那霉素、氯霉素、TaqDNA聚合酶、PfuDNA 聚合酶、dNTP Mixture、SanPrep柱式質(zhì)粒DNA小量抽提試劑盒等購自生工生物工程(上海)股份有限公司;DNA限制性內(nèi)切酶,DNA Marker,購自寶生物公司(中國,大連)。
主要儀器包括培養(yǎng)箱、恒溫?fù)u床、分光光度計(jì)、電轉(zhuǎn)儀、PCR儀、電泳儀、冷凍離心機(jī)、氨基酸分析儀等。
1.1.3 培養(yǎng)基及培養(yǎng)條件
LB培養(yǎng)基(胰蛋白胨10 g/L,酵母提取物5 g/L,NaCl 10 g/L,調(diào)pH至 7.0~7.2。固體培養(yǎng)基另加1.5%的瓊脂粉。)用于培養(yǎng)E.coliBL21(DE3)。培養(yǎng)溫度37 ℃。發(fā)酵培養(yǎng)基(葡萄糖10 g/L,甘油5 g/L,胰蛋白胨15 g/L,(NH4)2SO45 g/L,K2HPO43 g/L,F(xiàn)eSO43 mmol/L,MgSO40.2 g/L,CaCl20.015 g/L)用于反式-4-羥脯氨酸發(fā)酵,用NaOH調(diào)節(jié) pH 至8.0,250 mL三角錐形瓶分裝30 mL。菌株E.coliBL21(DE3)ΔputA/pUC19-proB2A-Ptrp2-hyp發(fā)酵生產(chǎn)時(shí),需要添加6~10 g/LL-脯氨酸;E.coliBL21(DE3)ΔputAΔargB/pUC19-proB2A-Ptrp2-hyp初步發(fā)酵添加1 g/LL-精氨酸。
抗生素的工作質(zhì)量濃度:氨芐青霉素50 μg/mL,卡那霉素50 μg/mL,氯霉素50 μg/mL。
1.1.4 引物設(shè)計(jì)
本實(shí)驗(yàn)所用引物序列見表2,由上海生工合成。

表2 實(shí)驗(yàn)所用引物
注:下劃線部分是質(zhì)粒pKD4的抗性基因序列。
1.2 重組大腸桿菌的構(gòu)建
1.2.1 敲除基因argB獲得精氨酸缺陷型菌株
利用Red/ET同源重組系統(tǒng)[11-13]進(jìn)行基因敲除(圖2)。
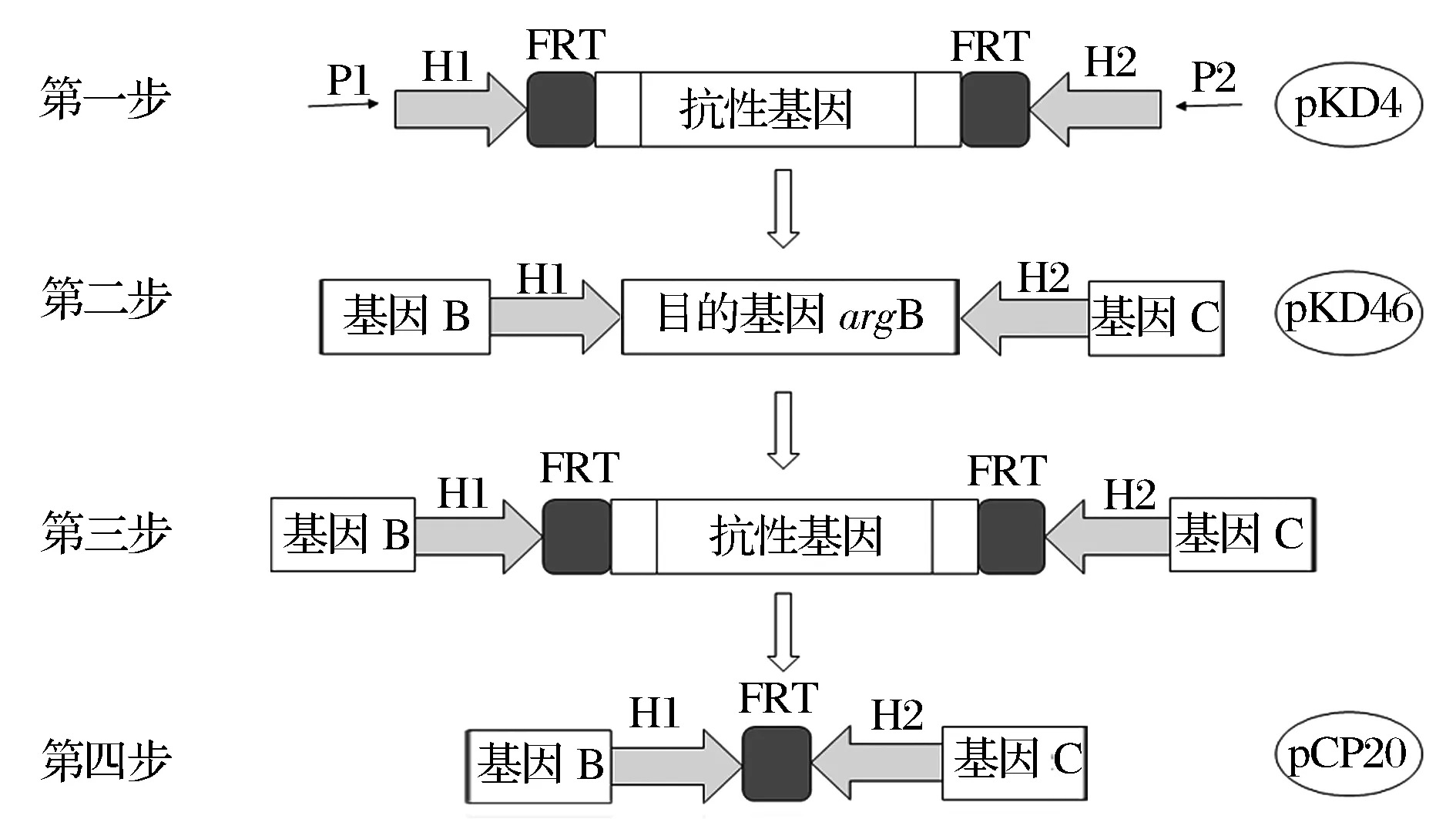
H2, H1是同源臂;P1, P2是抗性基因序列圖2 利用Red/ET同源重組系統(tǒng)進(jìn)行基因敲除的過程Fig.2 Processes of gene knockout using the plasmid-assistant Red homologous recombination
以輔助質(zhì)粒pKD4為模板,利用引物P1、P2通過PCR擴(kuò)增直接獲得含有目的基因上下游同源臂和kan基因的線性打靶片段;將表達(dá)Red重組酶的輔助質(zhì)粒pKD46導(dǎo)入E.coliBL21(DE3)ΔputA感受態(tài)細(xì)胞內(nèi),并制備為電轉(zhuǎn)感受態(tài)細(xì)胞(制備過程中需要添加L-阿拉伯糖以誘導(dǎo)質(zhì)粒pKD46的Exo蛋白、Beta蛋白和Gam蛋白的表達(dá));將線性的打靶片段電轉(zhuǎn)導(dǎo)入電轉(zhuǎn)感受態(tài)細(xì)胞中以實(shí)現(xiàn)打靶片段與目的基因argB的配對(duì)重組(以kan基因片段替代目的基因argB),之后采用42 ℃熱激耦合抗生素抗性負(fù)篩選的策略去除質(zhì)粒pKD46;通過菌落PCR對(duì)在卡那抗性平板上篩到的單菌落進(jìn)行驗(yàn)證;將表達(dá)FLP重組酶的輔助質(zhì)粒pCP20導(dǎo)入菌落PCR驗(yàn)證正確的陽性轉(zhuǎn)化子中,F(xiàn)LP重組酶可以直接作用于兩個(gè)FRT位點(diǎn)并發(fā)生同源重組,去除2個(gè)FRT位點(diǎn)之間的kan基因片段以消除卡那抗性,并留下一個(gè)FRT“疤痕”[14]。
1.2.2 大腸桿菌電轉(zhuǎn)感受態(tài)制備方法
挑取含有pkD46的E.coliBL21(DE3)ΔputA單菌落接種于氨芐抗性的新鮮LB液體培養(yǎng)基中,30 ℃,220 r/min過夜培養(yǎng);取過夜種子液按1%接種量接種到50 mL 氨芐抗性的新LB培養(yǎng)基中,并加入4.8 g/L的L-阿拉伯糖,30 ℃,220 r/min振蕩培養(yǎng)至OD600值達(dá)到0.5~0.6;將培養(yǎng)液分裝于滅菌的50 mL離心管中,冰上預(yù)冷 10 min,4 ℃,4 000 r/min離心10 min,棄去上清液,回收細(xì)胞;加入等體積的冰預(yù)冷10%(v/v)甘油重懸,4 ℃,4 000 r/min離心10 min,重復(fù)甘油洗2次;小心棄去上清,利用殘留的甘油重懸細(xì)胞,每管分裝80 μL,-80 ℃保存。
1.2.3 大腸桿菌化轉(zhuǎn)感受態(tài)制備方法
接種大腸桿菌單菌落于50 mL pH 7.0的新鮮LB液體培養(yǎng)基中,30 ℃,220 r/min過夜培養(yǎng);取過夜種子液按1%接種量接種到pH 7.0的新鮮LB培養(yǎng)基中,30 ℃,220 r/min振蕩培養(yǎng)(約2 h)至 OD600值達(dá)到0.5~0.6;將培養(yǎng)液分裝于滅菌的50 mL離心管中,冰上預(yù)冷 10 min;4 ℃,1 000×g 離心5 min,棄去上清,回收細(xì)胞;加入原培養(yǎng)液5%體積的TSB培養(yǎng)基充分懸浮菌體,緩慢吹打均勻;冰上放置10 min后分裝至已滅菌的1.5 mL離心管中,每管分裝50 μL,-80 ℃保存。
1.2.4 大腸桿菌感受態(tài)細(xì)胞的轉(zhuǎn)化方法(熱擊法)[4]
轉(zhuǎn)化體系為:30 μL ddH2O,10 μL 5×KCM(5 mol/L KCl,0.15 mol/L CaCl2,0.25 mol/L MgCl2),10 μL重組質(zhì)粒,10 μL感受態(tài)細(xì)胞。迅速混勻,先冰浴30 min;然后42 ℃熱擊90 s;接著冰浴5 min;最后加入600 μL新鮮的LB液體培養(yǎng)基,37 ℃ 220 r/min振蕩培養(yǎng)1 h;取100 μL復(fù)蘇培養(yǎng)后的菌液涂布于含抗生素的LB平板,于37 ℃恒溫培養(yǎng)箱中培養(yǎng)8~16 h。
1.3 發(fā)酵試驗(yàn)
1.3.1 搖瓶發(fā)酵
將重組大腸桿菌E.coliBL21(DE3)ΔputAΔargB/ pUC19-BH在含有氨芐霉素的LB平板上劃線培養(yǎng);挑取單菌落接種于含有50 μg/mL 氨芐的LB液體培養(yǎng)基中(30 mL/250 mL三角瓶),37 ℃ 220 r/min振蕩培養(yǎng)8 h,作為種子液;按6%接種量取上述種子培養(yǎng)液1.8 mL接入發(fā)酵培養(yǎng)基(30 mL/250 mL三角瓶),30 ℃ 220 r/min振蕩培養(yǎng)24 h。
1.3.2 生物量的測(cè)定
取發(fā)酵液,用去離子水稀釋相應(yīng)倍數(shù),并以去離子水為空白對(duì)照,測(cè)定600 nm波長(zhǎng)處的吸光度值。
1.3.3 葡萄糖的測(cè)定
取1 mL發(fā)酵液,8 000 r/min 離心2 min,取200 μL上清液,加入200 μL DNS試劑,沸水浴5 min;用冷水冷卻樣品,然后加2.6 mL去離子水稀釋到3 mL,用分光光度計(jì)在540 nm波長(zhǎng)處測(cè)定吸光度值。
1.3.4L-脯氨酸的含量測(cè)定
L-脯氨酸的檢測(cè)采用酸性茚三酮顯色法:將發(fā)酵液離心后,取上清,適當(dāng)稀釋后取1 mL于10 mL試管中,加入1 mL冰乙酸;搖勻后加入1 mL酸性茚三酮;搖勻后沸水浴1 h;接著加入冰乙酸2 mL,搖勻后冷水冷卻,測(cè)定515 nm波長(zhǎng)處的吸光度值。
1.3.5 反式-4-羥脯氨酸積累量的測(cè)定
反式-4-羥脯氨酸的檢測(cè)采用氯胺T法:將發(fā)酵液離心后,取上清,稀釋后取2.5 mL于10 mL試管中,加入1 mL氯胺T,搖勻后室溫放置20 min;然后加入1 mL顯色劑,搖勻后迅速將試管置于60 ℃水浴鍋中,20 min后取出冷水冷卻,用分光光度計(jì)在560 nm波長(zhǎng)處測(cè)定吸光度值。
1.3.6 反式-4-羥化酶酶活的測(cè)定[15]
全細(xì)胞酶活測(cè)定條件:測(cè)定發(fā)酵液的OD600,根據(jù)大腸桿菌OD600和細(xì)胞干重的常用轉(zhuǎn)換計(jì)算公式,根據(jù)DCW(g/L) =0.54×OD600測(cè)得細(xì)胞干重DCW。發(fā)酵液12 000 r/min離心2 min后,棄掉上清液,回收菌體,將500 μL的酶反應(yīng)緩沖液(MES 240 mmol/L(pH 6.5),脯氨酸20 mmol/L,2-酮戊二酸40 mmol/L,硫酸亞鐵4 mmol/L,維生素C 8 mmol/L)加入到稱重好的菌體中,用酶反應(yīng)緩沖液將細(xì)胞重新懸浮后,在35 ℃搖床中,220 r/mim培養(yǎng)15 min后,轉(zhuǎn)移到100 ℃水浴中加熱2 min終止酶反應(yīng),并測(cè)定反式-4-羥脯氨酸濃度,反式-4-羥脯氨酸的測(cè)定方法同上1.3.5。1個(gè)單位酶活定義1 min將1 nmol脯氨酸完全轉(zhuǎn)化為反式-4-羥脯氨酸的酶量,單位為U,全細(xì)胞酶活是每1 mg干菌體的酶活,單位為U/mg。
1.3.7 乙酰谷氨酸激酶活力測(cè)定[16]
2 mL反應(yīng)總體積中,含0.1 mL 酶液,0.1 mL 0.2 mol/L鹽酸羥胺,0.28 mL 0.14 mol N-乙酰谷氨酸,0.2 mL 0.02 mol MgCl2,0.14 mL 0.014 mol ATP,1.18 mL 0.1 mol Tris-HCl緩沖液(pH8.0),37 ℃水浴保溫30 min,加2 mL FeCl3試劑(3.4%FeCl3溶于2 mol/L HCl,并與8%三氯乙酸等體積混勻)終止反應(yīng),測(cè)OD540值。1個(gè)酶活力單位(U)是指在上述條件下,反應(yīng)體系中每分鐘催化1 μmol N-乙酰谷氨酸氧化所需的酶量。
1.3.8 氨基酸的測(cè)定
發(fā)酵液經(jīng)12 000 r/min 離心5 min 后,取上清500 μL,加入500 μL 10%磺基水楊酸沉淀4 h,用0.02 mol/L HCl稀釋適當(dāng)倍數(shù),用0. 22 μm微孔膜過濾后,采用氨基酸分析儀 L-8900測(cè)定氨基酸。
2 結(jié)果與分析
2.1argB敲除菌株的驗(yàn)證
經(jīng)過抗性平板篩選,挑出只在非抗性平板上菌落,用驗(yàn)證引物Y1、Y2進(jìn)行菌落PCR,并進(jìn)行核酸膠驗(yàn)證。篩選到argB成功敲出的菌株。從圖3中可以看到出發(fā)菌株E.coliBL21(DE3)ΔputA菌落PCR得到片段大小為1 000 bp,與通過驗(yàn)證引物Y1、Y2進(jìn)行菌落PCR獲得的片段大小相當(dāng)(片段大小為964 bp);卡那片段成功替換菌株菌落PCR得到片段大小為1 700 bp左右,與卡那基因片段大小相當(dāng);消抗后從圖3可以看到除了菌株3-8,其余15株菌經(jīng)過菌落PCR得到大小為300 bp左右的片段,說明argB基因被成功敲除。由于利用Red/ET同源重組系統(tǒng)進(jìn)行基因敲除會(huì)留下一個(gè)FRT“疤痕”,可能會(huì)對(duì)菌株生長(zhǎng)造成影響,因此,本實(shí)驗(yàn)對(duì)該15株菌進(jìn)行了初步發(fā)酵,發(fā)現(xiàn)菌株3-4脯氨酸產(chǎn)量高于其他菌株,且高于菌株BL21(DE3)ΔputA(圖4),得到菌株BL21(DE3)ΔputAΔargB。
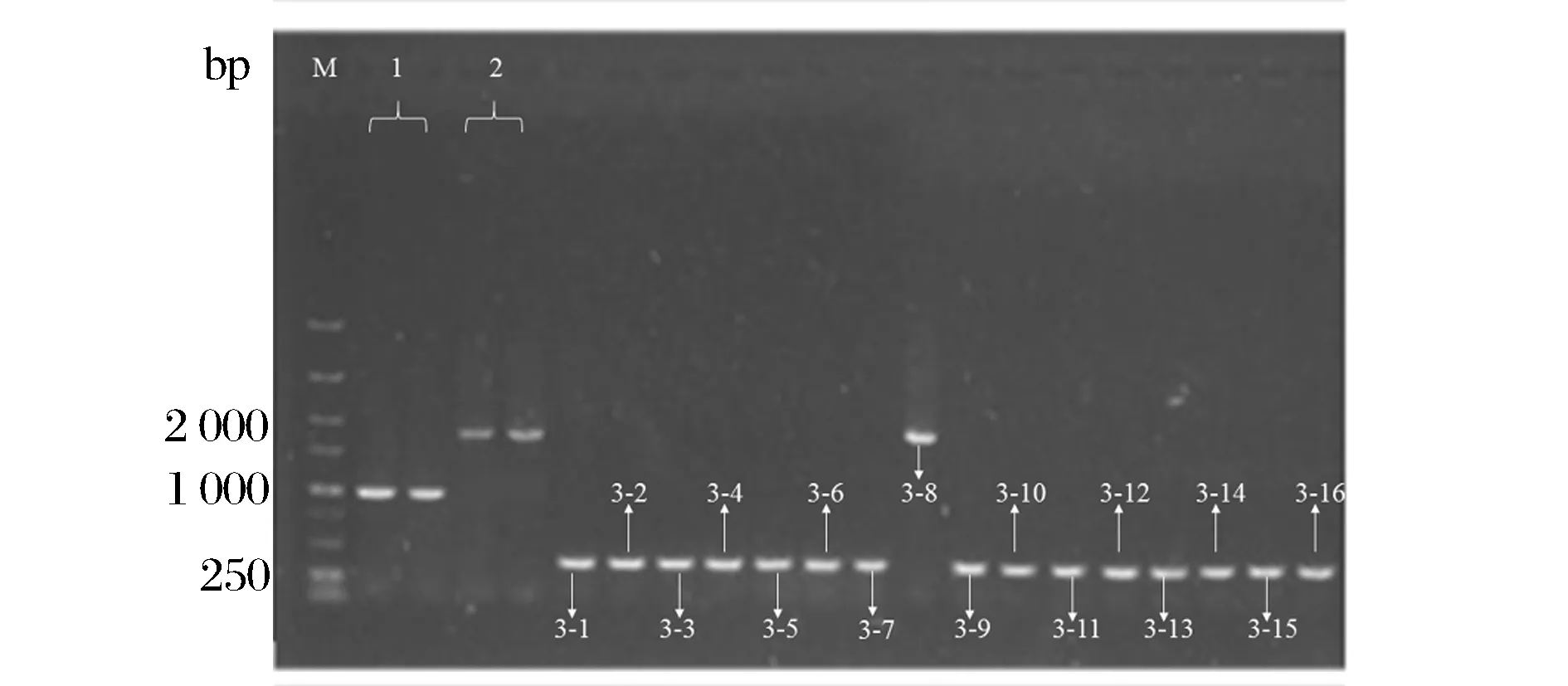
M-5 000 bp DNA Ladder Marker;1-E. coliBL21(DE3) ΔputA的菌落PCR結(jié)果;2-E. coli BL21(DE3) ΔputA抗性替換成功菌株的菌落PCR結(jié)果;3-抗性消除菌株的菌落PCR結(jié)果圖3 argB基因敲除菌株的PCR鑒定Fig.3 PCR identification of argB gene knockout strain

1代表菌株BL21(DE3)ΔputA的初步發(fā)酵結(jié)果;2、3、4、5、6、7、8、9、10、11、12、13、14、15、16分別代表目標(biāo)菌株 3-1、3-2、3-3、3-4、3-5、3-6、3-7、3-9、3-10、3-11、3-12、3-13、3-14、3-15、3-16的初步發(fā)酵結(jié)果圖4 argB基因敲除菌株的初步發(fā)酵Fig.4 The preliminary fermentation of argB gene knockout strain
2.2 重組菌株的構(gòu)建
通過材料與方法1.2.4中化學(xué)轉(zhuǎn)化的方法,構(gòu)建菌株BL21(DE3)ΔputAΔargB/pUC19-BH。通過使用SanPrep柱式質(zhì)粒DNA小量抽提試劑盒提取重組菌株BL21(DE3)ΔputAΔargB/pUC19-BH。質(zhì)粒進(jìn)行單雙酶切驗(yàn)證(圖5)。質(zhì)粒通過BamH I、KpnI進(jìn)行雙酶切,分別獲得基因片段hyp-Ptrp2-proB2A(BH片段,3416 bp)、載體pUC19(2 686 bp),結(jié)果如泳道1;表達(dá)載體通過BamH I進(jìn)行單酶切,重組質(zhì)粒的大小約6 102 bp,結(jié)果如泳道2;從圖5可以看出這些片段的大小均與預(yù)期目標(biāo)相符,表明重組菌株構(gòu)建成功。

M1-5000bp DNA Ladder Marker;M2-5000bp DNA Ladder Marker;1-雙酶切結(jié)果;2-單酶切結(jié)果圖5 表達(dá)質(zhì)粒酶切電泳驗(yàn)證Fig.5 The electrophoresis test and verify of recombinant plasmid pUC19-BH by enzyme digestion
2.3 氨基酸分析儀檢測(cè)發(fā)酵液中氨基酸成分
通過氨基酸分析儀進(jìn)行初步定性分析結(jié)果(圖6),可以發(fā)現(xiàn)菌株BL21(DE3)ΔputA/pUC19-BH與菌株BL21(DE3)ΔputAΔargB/pUC19-BH發(fā)酵結(jié)果相比,前者精氨酸的含量明顯高于后者,精氨酸缺陷型菌株幾乎沒有精氨酸積累,這進(jìn)一步證明后者精氨酸途徑被成功切斷。同時(shí),我們看到發(fā)酵液中有反式-4-羥脯氨酸生成,說明羥化酶得到表達(dá),菌株BL21(DE3)ΔputAΔargB/pUC19-BH構(gòu)建成功。

圖6 發(fā)酵液中各氨基酸分析結(jié)果Fig.6 The analytical results of amino acids in fermentation broth圖a-1,a-2:菌株BL21(DE3)ΔputA/pUC19-BH發(fā)酵液中氨基酸分析結(jié)果;圖b-1,b-2:無外源添加L-精氨酸時(shí),菌株BL21(DE3)ΔputAΔargB/pUC19-BH發(fā)酵液中氨基酸分析結(jié)果;圖c-1,c-2:外源添加適量L-精氨酸時(shí),菌株BL21(DE3)ΔputAΔargB/pUC19-BH與發(fā)酵液中氨基酸分析結(jié)果
2.4argB基因敲除菌株發(fā)酵參數(shù)
對(duì)敲除菌進(jìn)行初步發(fā)酵,并測(cè)定菌株的發(fā)酵參數(shù),從結(jié)果(圖7)中,我們發(fā)現(xiàn)精氨酸缺陷型菌株無論是生長(zhǎng)狀況,葡萄糖消耗還是產(chǎn)反式-4-羥脯氨酸的能力均受到一定抑制。對(duì)于argB基因敲除菌株,該基因編碼的N-乙酰谷氨酸激酶的酶活幾乎為0 mU/mg,這更進(jìn)一步說明argB基因被成功敲除。另外,研究發(fā)現(xiàn),當(dāng)培養(yǎng)基中外源添加1 g/L的L-精氨酸時(shí),菌體的生長(zhǎng)狀況得到了恢復(fù),且反式-4-羥脯氨酸的產(chǎn)量與出發(fā)菌株相比有所提高。因此,后文L-我們對(duì)精氨酸的添加量進(jìn)行了研究。

a-菌株BL21(DE3)ΔputA/pUC19-BH的發(fā)酵參數(shù);b-無外源添加L-精氨酸時(shí),菌株 BL21(DE3)ΔputAΔargB/pUC19-BH的發(fā)酵參數(shù);c-外源添加1 g/L L-精氨酸時(shí),菌株BL21(DE3)ΔputAΔargB/pUC19-BH的發(fā)酵參數(shù)圖7 argB基因敲除菌株發(fā)酵結(jié)果Fig.7 The fermentationresults of argB knockout strains
2.5L-精氨酸對(duì)BL21(DE3)ΔputAΔargB/pUC19-BH反式-4-羥脯氨酸產(chǎn)量的影響
由于菌株是L-精氨酸營養(yǎng)缺陷型,所以種子培養(yǎng)基中需要加入適量的精氨酸來滿足菌體生長(zhǎng)的需求。本實(shí)驗(yàn)研究了精氨酸不同濃度對(duì)菌體的生長(zhǎng)。
argB基因的敲除,使得精氨酸合成受阻,菌株變?yōu)長(zhǎng)-精氨酸缺陷型菌株,因此外源添加精氨酸,可以補(bǔ)充菌體生命活動(dòng)所需的物質(zhì)。BL21(DE3)ΔputAΔargB/pUC19-BH菌體生長(zhǎng)能力的恢復(fù),有利于反式-4-羥脯氨酸產(chǎn)量的進(jìn)一步提高。發(fā)酵培養(yǎng)基中添加不同濃度的精氨酸,觀察菌體的生長(zhǎng)及反式-4-羥脯氨酸產(chǎn)量情況,結(jié)果如圖8所示。隨著精氨酸濃度的增加,菌株的生長(zhǎng)及反式-4-羥脯氨酸合成能力均有所提高。當(dāng)發(fā)酵培養(yǎng)基中添加的L-精氨酸濃度達(dá)到600 mg/L后,隨著精氨酸添加量的增加,反式-4-羥脯氨酸產(chǎn)量增長(zhǎng)趨勢(shì)變小。從生產(chǎn)成本考慮,選擇在培養(yǎng)基中添加600 mg/L的L-精氨酸,此時(shí)反式-4-羥脯氨酸產(chǎn)量達(dá)到 312.67 mg/L,較出發(fā)菌株BL21(DE3)ΔputA/pUC19-BH產(chǎn)量233.59 mg/L提高了25.29% ;細(xì)胞生長(zhǎng)量也恢復(fù)到了親本水平。由此可見,精氨酸的添加有利于菌株的生長(zhǎng)及反式-4-羥脯氨酸的積累。

圖8 L-精氨酸添加量的優(yōu)化Fig.8 The optimization of addition amount of L-arginine
3 討論
本研究通過Red/ET同源重組系統(tǒng)進(jìn)行基因敲除,成功構(gòu)建了argB基因敲除菌株 BL21(DE3)ΔputAΔargB,結(jié)果發(fā)現(xiàn)該重組菌株的反式-4-羥脯氨酸產(chǎn)量有所提高。說明argB基因的敲除有利于反式-4-羥脯氨酸的生產(chǎn)。
argB基因編碼精氨酸合成途徑的乙酰谷氨酸激酶。敲除argB基因后,胞內(nèi)精氨酸合成受阻,可能使更多前體谷氨酸流向L-脯氨酸合成途徑,從而提高作為反式-4-羥脯氨酸底物的L-脯氨酸產(chǎn)量。但L-精氨酸是必須氨基酸,對(duì)維持菌體正常生命活動(dòng)有重要意義,所以其敲除影響了菌體的生長(zhǎng)。我們對(duì)L-精氨酸的添加量進(jìn)行了研究,結(jié)果發(fā)現(xiàn)適量的精氨酸添加可以使得敲除菌株恢復(fù)生長(zhǎng)性狀,同時(shí)反式-4-羥脯氨酸的產(chǎn)量達(dá)到312.67 mg/L與實(shí)驗(yàn)室原有水平相比有了顯著的提高。
該生產(chǎn)反式-4-羥脯氨酸的方法與原生產(chǎn)方法相比,原生產(chǎn)法方在搖瓶發(fā)酵時(shí)需要添加6~10 g/L的L-脯氨酸作為底物,而該生產(chǎn)方法只需添加0.6~1 g/L的L-精氨酸來滿足菌體生長(zhǎng);且L-脯氨酸的價(jià)格高于L-精氨酸。因此本文所構(gòu)建的菌株在生產(chǎn)過程中展現(xiàn)出成本低,發(fā)酵過程易控制,發(fā)酵水平穩(wěn)定等優(yōu)勢(shì),這為反式-4-羥脯氨酸的工業(yè)化生產(chǎn)奠定了良好基礎(chǔ)。
[1] VICKERY H B, SCHMIDT C L A. The history of the discovery of the amino acids[J]. Chemical Reviews,1931,9(2): 169-318.
[2] BRANDS K M J, JOBSON R B, CONRAD K M, et al. Efficient one-pot synthesis of the 2-aminocarbonylpyrrolidin-4-ylthio-containing side chain of the new broad-spectrum carbapenem antibioticertapenem[J].The Journal of Organic Chemistry,2002,67(14): 47 71-4 776.
[3] PHANG J M, DONALD S P, PANDHARE J, et al. The metabolism of proline, a stress substrate, modulates carcinogenic pathways[J]. Amino Acids, 2008, 35(4): 681-690.
[4] 劉合棟. 高產(chǎn)反式-4-羥脯氨酸重組大腸桿菌的構(gòu)建和發(fā)酵優(yōu)化[D]. 無錫: 江南大學(xué), 2013.
[5] 張勝利.產(chǎn)反式-4-羥基-L-脯氨酸大腸桿菌菌株的改造及發(fā)酵條件初步研究[D]. 無錫: 江南大學(xué), 2015.
[6] 許虹, 竇文芳, 許泓瑜,等.不同供氧水平對(duì)L-精氨酸分批發(fā)酵過程的影響[J]. 化工學(xué)報(bào), 2008, 59(9):2 295-2 301.
[7] 陳曉博, 羅雪粵, 張淑榮,等. 生產(chǎn)L-精氨酸的脯氨酸營養(yǎng)缺陷型菌株的選育[J]. 北京化工大學(xué)學(xué)報(bào)(自然科學(xué)版) , 2011, 38(6): 83-86.
[8] LEE S Y, PARK J M, LEE J H, et al. Interaction of transcriptional repressor ArgR with transcriptional regulator FarR at theargBpromoter region inCorynebacteriumglutamicum[J]. Ashaipplied and Environmental Microbiology, 2011, 77(3): 711-718.
[9] SOO Y L, HWA S S , PARK J S, et al.Proline reduces the binding of transcriptional regulator ArgR to upstream ofargBinCorynebacteriumglutamicum[J]. Applied Genetics AndMolecular Biotechnology, 2010, 86(1): 235-242.
[10] 胡丹丹.無外源L-脯氨酸發(fā)酵生產(chǎn)反式-4-羥脯氨酸重組大腸桿菌的構(gòu)建和發(fā)酵優(yōu)化[D]. 無錫: 江南大學(xué), 2015.
[11] DATSENKO K A, WANNER B L. One-step inactivation of chromosomal genes inEscherichiacoliK-12 using PCR products[J]. Proceedings of the National Academy of Sciences of the United States of America, 2000, 97(12): 6 640-6 645.
[12] POTEETE A R, FENTON A C. Genetic requirements of phage lambda red-mediated gene replacement inEscherichiacoliK-12[J]. Journal of Bacteriology, 2000, 182(8): 2 336-2 340.
[13] BABA T, ARA T, HASEGAWA M, et al. Construction ofEscherichiacoliK-12 in-frame, single-gene knockout mutants: the Keio collection[J]. Molecular Systems Biology, 2006,2(1): 1-11.
[14] DOUBLET B, DOUARD G,TARGANT H, et al. Antibiotic marker modifications of λ Red and FLP helper plasmids, pKD46 and pCP20,for inactivation of chromosomal genes using PCR products in multidrug-resistant strains[J]. Journal of Microbiological Methods, 2008, 75 (2): 359-361.
[15] SHIBASAKI T, OZAKI A, MORI H. Enzymatic production oftrans-4-hydroxy-L-proline by regio- and stereospecific hydroxylation ofL-proline[J]. Bioscience Biotechnology & Biochemistry,2000, 64(4): 746-750.
[16] 陳雪嵐, 許正宏, 陶文沂. 鈍齒棒桿菌產(chǎn)精氨酸關(guān)鍵酶分析[J]. 食品科學(xué), 2005, 26(3): 35-38.
Fermentative production oftrans-4-hydroxyproline with arginine-deficient strain
WANG Xiao-jiao,ZHANG Zhen-yu*,SUN Fu-bao*,YU Lin
(College of Bioengineering, Jiangnan University, Wuxi 214122, China)
In order to obtain high-yield strains oftrans-4-hydroxyproline, based on the guidance ofE.colimetabolic network model and usingE.coliBL21 (DE3)ΔputAas the original strain, geneargBwas knocked out successfully, which blocked metabolic pathways branch ofL-glutamic acid,L-proline synthetic precursors and increased synthesis ofL-proline metabolic flux. The constructed arginine-deficient strainE.coliBL21 (DE3)ΔputAΔargBwas transformed with the expression plasmid pUC19-proB2A-Ptrp2-hyp. The expression plasmid contains the mutant geneproB2encoding glutamate kinase that significantly reducedL-proline feedback inhibition. Fermentation results indicated thatthetrans-4-hydroxyproline production of recombinant strain reached 312.67 mg/L after addition of 600 mg/L exogenous arginine, which increased by 25.29% compared withE.coliBL21(DE3)ΔputA/ pUC19-proB2A-Ptrp2-hyp.
Escherichiacoli;trans-4-hydroxyproline; knockout; proline
10.13995/j.cnki.11-1802/ts.201701005
碩士研究生(張震宇教授,孫付保副教授為通訊作者,E-mail:zhangzy@gmail.com;fubaosun@jiangnan.edu.cn)。
國家自然科學(xué)基金(30970058);江蘇省自然科學(xué)基金(BK2012554);工業(yè)生物技術(shù)教育部重點(diǎn)實(shí)驗(yàn)室(江南大學(xué))開放課題基金(KLIB-ZR200801)
2016-07-18,改回日期:2016-09-09
