海藍組織細胞增生癥1例
2017-04-14 11:17:11陳萼珊
臨床與實驗病理學雜志 2017年2期
陳萼珊,劉 偉
海藍組織細胞增生癥1例
陳萼珊1,2,劉 偉1
海藍組織細胞增生癥;骨髓;病例報道
患者男性,19歲,因頸部淋巴結腫痛4天、發熱1天就診。查體:全身皮膚無黃染,無出血點、瘀點、瘀斑。頸前、頜下、頦下、枕后多發淺表淋巴結腫大,疼痛明顯,拒按,表面無紅腫、破潰、出血,余淺表淋巴結未觸及腫大。心肺腹及四肢反射未見明顯異常。實驗室檢查:血常規WBC 9.79×109/L,ANC 0.53×109/L,HB 122 g/L,PLT 37×109/L;血生化:丙氨酸轉氨酶201.5 U/L、谷草轉氨酶128.0 U/L、乳酸脫氫酶928.0 U/L、α羥丁酸脫氫酶697.0 U/L;凝血功能:D-二聚體1.43 mg/L,凝血酶原時間14.3 s,PT-INR 1.23,凝血酶時間23.6 s;C-反應蛋白16.7 mg/L;余項未見明顯異常。遂行骨髓活檢術。
病理檢查 眼觀:灰黃色穿刺組織1條,長1.3 cm,直徑0.3 cm。鏡檢:骨髓腔3個,造血成分占60%,脂肪占40%,三系細胞存在,粒系約占70%,有核紅細胞約占10%,造血組織中可見較多組織細胞浸潤,胞質內含棕黃色顆粒(圖1)。免疫表型:組織細胞CD163陽性(圖2),胞質內顆粒呈PAS、D-PAS及PAM染色陽性(圖3)。骨髓涂片:有核細胞增生活躍,粒系占63.5%,紅系占16%,粒系紅系形態大致正常,淋巴細胞占16%,巨核細胞165個,血小板少見,原始粒細胞占5%,海藍細胞常見(圖4)。
病理診斷:海藍組織細胞增生癥。
討論 海藍組織細胞增生癥亦稱海藍組織細胞綜合征(sea blue histiocyte syndrome, SBH),由Sawitsky等[1]于1954年首次報道,1970年由Silverstein[2]正式命名。目前認為SBH是一種脂質代謝異常疾病,其本質為類脂質分解代謝途徑中某種酶的遺傳性缺陷或過度負荷以致代謝的中間產物(泡沫細胞內蠟樣物質或神經鞘糖磷脂)在體內儲積并被單核巨噬細胞吞噬,出現吞噬這類脂質物質的組織細胞增生,并浸潤各臟器引起相應的臨床癥狀和體征的一類疾病。也有學者[3]認為SBH可能是尼曼皮克病一種變異的類型。
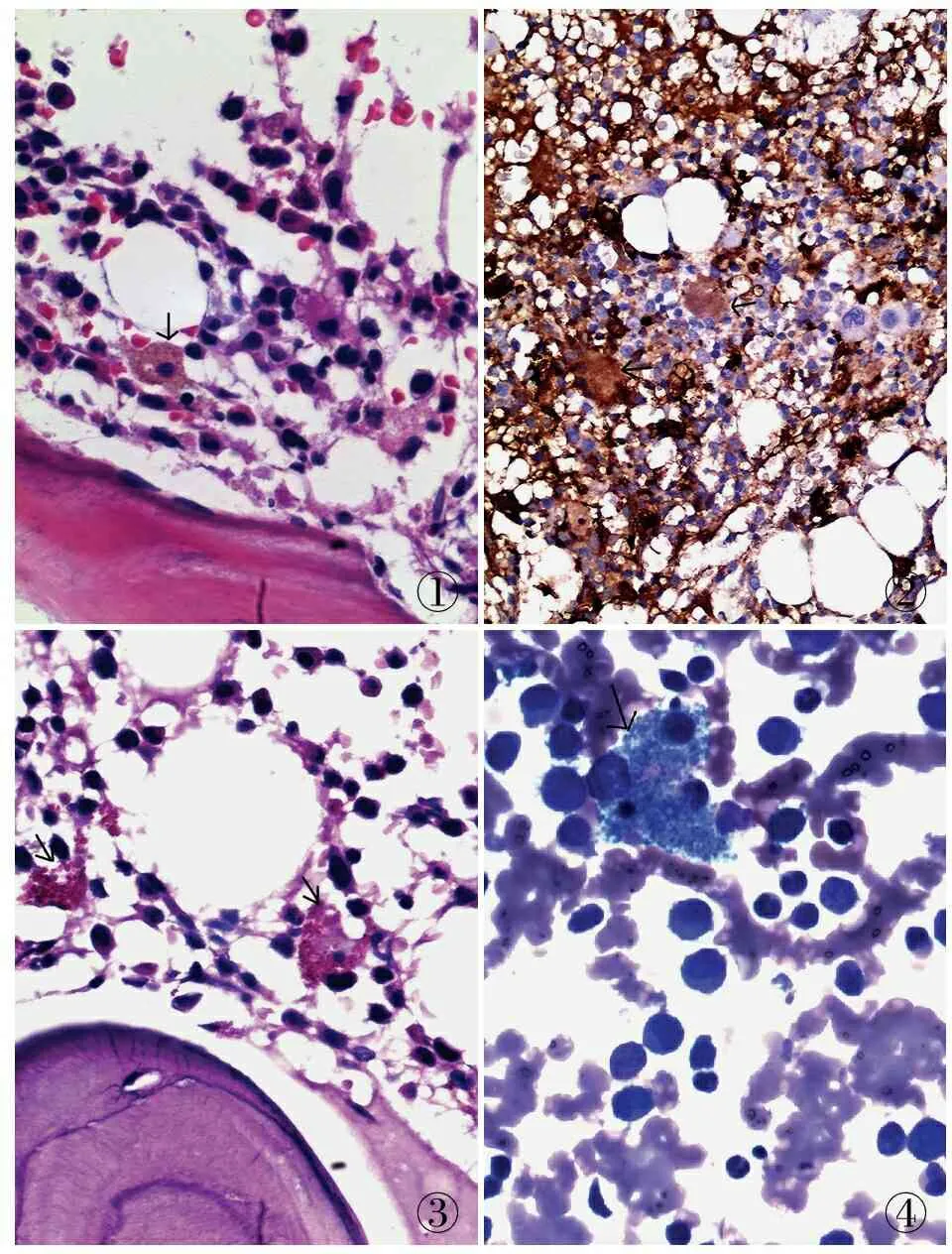
圖1 造血組織中可見組織細胞浸潤,胞質內含棕黃色顆粒 圖2 組織細胞CD163陽性,SP法 圖3 胞質內顆粒PAS染色陽性,SP法 圖4 骨髓涂片中可見海藍細胞
SBH分為原發性、原發性家族性和繼發性三種;原發性SBH少見,可能為先天性缺乏鞘磷脂酶所致,需先排除家族性及繼發性。原發性家族性SBH可能與APO基因突變相關,家系調查顯示為常染色體隱性遺傳病。繼發性SBH目前報道較多,可能與該類患者酶異常、脂質代謝負擔過重或細胞代謝增加有關,可繼發于多種疾病,主要包括慢性粒細胞白血病、骨髓增生異常綜合征、淋巴瘤等淋巴造血系統疾病,Turner綜合征、肝硬化、甲亢、脾亢,以及接受鎮靜劑、抗風濕、抗白血病、anorexic和兩性霉素B[4]等藥物治療及全腸外營養后。
SBH可見于任何年齡,無性別差異,臨床表現以肝脾腫大、血小板減少、貧血最常見,亦有以皮膚肌肉[5]改變為首發癥狀的報道。若SBH合并腦損害時可出現進行性神經系統受累,肺部損害可類似肺纖維化或結核性肉芽腫甚至粟粒樣肺結核浸潤現象,累及腎臟可引起血尿及蛋白尿。本例患者臨床特點主要為淺表淋巴結腫大、血小板減少及轉氨酶升高,無其他繼發性疾病,家族中無類似患者,骨髓涂片及骨髓活檢發現海藍組織細胞,故支持原發性SBH診斷。
SBH確診主要依據骨髓或原發病灶內病理檢查發現海藍組織細胞,部分繼發性骨髓外SBH可通過局部組織穿刺細胞學查見海藍細胞進行確診,如額骨腫塊、頜竇囊腫、甲狀腺腫、胸壁包塊、子宮頸、乳腺溢液、心包積液等。本例行骨髓活檢標本HE染色顯示鏡下海藍組織細胞呈棕黃色,胞體較大,外形不規則,胞質較豐富,呈粗顆粒狀、空泡網狀,核染紫黑色,大小不一,呈不規則圓形或粗糙塊狀,多偏位;PAS、PAM染色陽性,可與組織細胞吞噬含鐵血黃素鑒別。
鑒別診斷:(1)尼曼皮克:以神經鞘磷脂沉積引起的一類常染色體隱性遺傳性疾病,臨床表現主要是肝脾腫大、眼底櫻桃樣紅斑。活檢見尼曼-匹克細胞,細胞胞質淺染,泡沫狀,有大核仁。(2)戈謝病:臨床主要有肝脾大、眼球結膜黃斑及骨病變,血清酸性磷酸酶升高,活檢標本中Gaucher細胞胞體大,胞質豐富,淡染,油鏡下可見胞質充滿皺紋紙樣“絲狀物質”。(3)繼發性SBH:有原發病的相關臨床特征,易于鑒別。
本病尚無特殊治療方法,有脾大或脾亢者可行脾切除術,但僅能緩解癥狀,不能解除病因,且手術治療還有可能加速高脂血癥和肝臟脂質沉積[5]。部分繼發性骨髓外SBH清除原發病灶后可痊愈[6]。大多數預后呈良性病程,年齡越小預后越差,約15%患者死于肝硬化[7],也可因胃腸道大出血或肺部病變而死亡。
[1] Sawitsky A, Hyman G A, Hyman J B. An unidentified reticuloendothelial cell in bone marrow and spleen; report of two cases with histochemical studies[J]. Blood, 1954,9(10):977-985.
[2] Silverstein M N, Ellefson R D, Ahern E J. The syndrome of the sea-blue histiocyte[J]. N Engl J Med, 1970,282(1):1-4.
[3] Gunay E, Firat Guyen S, Aktas Z,etal. Pulmonary involvement in sea-blue histiocytosis[J]. Tuberk Toraks, 2012,60(2):176-179.
[4] Michot J M, Gubavu C, Fourn E,etal. Very prolonged liposomal amphotericin B use leading to a lysosomal storage disease[J]. Int J Antimicrob Agents, 2014,43(6):566-569.
[5] 謝 易, 傅君芬, 梁 黎. 原發性海藍組織細胞增多癥1例[J]. 中國實用兒科雜志, 2013,28(1):74-75.
[6] 李繼廣, 王克強. 8例骨髓外局部組織檢出海藍組織細胞分析[J]. 中國實驗診斷學, 2008,12(12):1585.
[7] Suzuki O, Abe M. Secondary sea-blue histiocytosis derived from niemann-pick disease[J]. J Clin Exp Hematop, 2007,47(1):19-21.
時間:2017-2-27 10:14
http://kns.cnki.net/kcms/detail/34.1073.R.20170227.1016.074.html
1福州總醫院病理科,福州 3500252福州總醫院附屬第一醫院(解放軍第95醫院)病理科,莆田 351100
陳萼珊,女,醫師。E-mail: cessue@163.com 劉 偉,男,碩士,主治醫師,通訊作者。E-mail: liuweiyisong@163.com
R 589.2
B
1001-7399(2017)02-0230-02
10.13315/j.cnki.cjcep.2017.02.033
接受日期:2016-10-10
