桿狀體肌病臨床、病理及分子生物學研究進展
2017-11-03 12:39:25趙燕劉卓常蕾蕾盧正娟吳家勇牛豐南徐運
臨床神經病學雜志 2017年5期
趙燕,劉卓,常蕾蕾,盧正娟,吳家勇,牛豐南,徐運
·綜述·
桿狀體肌病臨床、病理及分子生物學研究進展
趙燕,劉卓,常蕾蕾,盧正娟,吳家勇,牛豐南,徐運
先天性肌病(CMs)是一組單基因遺傳性骨骼肌疾病,具有臨床/遺傳異質性,活檢骨骼肌病理存在特定的結構改變。遺傳方式有常染色體顯性遺傳(AD)、常染色體隱性遺傳(AR)、X連鎖隱性遺傳(XR)及散發。發病率暫無統一統計,瑞典估計約為1/22 480,北愛爾蘭為1/13 500[1]。典型臨床表現多數以新生兒/幼兒期發病的四肢/軸向肌群肌無力及肌張力低下,病情相對穩定/進展緩慢。可伴有面肌受累(細長臉、高顎弓等)和骨關節受累(先天性髖關節發育不良、脊柱側彎、跟腱攣縮等)。血清肌酸激酶(CK)正常/輕度升高,EMG正常/肌源性改變。骨骼肌活檢共同的病理特點:無明顯肌纖維變性、壞死,可存在肌纖維類型分布異常。根據其特征性病理特點,North等[2]將CMs主要分為以下5類:(1)軸空性肌病,包括中央軸空病(CCD)和多微小軸空病(MmD);(2)中心核肌病(CNMs);(3)桿狀體肌病(NM),根據桿狀體不同的病理表現形式還包括帽狀肌病、斑馬體肌病、軸空/桿狀體肌病;(4)肌球蛋白儲積病(MSM)又稱為透明體肌病;(5)先天性肌纖維類型不均(CFTD)。
NM是CMs中最常見的類型之一,呈AD/AR遺傳。NM具有廣泛的臨床表現,肌無力程度輕重不等,最嚴重的患兒在出生后無法自主呼吸;而癥狀較輕的患者僅有輕度肢體無力,病情穩定/不進展[3]。
1 臨床特點
根據ENMC國際協作組[4]的分型,NM主要分為以下6型:(1)先天性NM嚴重型(16.1%)[5],出生時即有嚴重的肌無力、肌張力低下,無自發運動及自主呼吸,很快死亡;(2)中間型(20.3%),較先天性NM嚴重型稍輕,肢體肌力可達Ⅰ級,通過呼吸支持可以維持生命,但不能行走;(3)輕癥型(即經典型)(46.1%),嬰兒期/兒童期起病,主要以面肌、軀干肌無力,伴肌張力低下,運動發育遲緩,病情相對穩定;(4)兒童起病嚴重型(13.3%);(5)散發的成人起病型(4.2%);(6)少數骨骼肌病理發現桿狀體,但需診斷為骨骼肌細肌絲蛋白病或肌病伴桿狀體,以區別先天性桿狀體肌病。根據患者癥狀的嚴重程度對NM進行分類,有助于判斷該病的預后及可能的致病基因。但隨著分子生物學的發展,特定基因變異對應特定的臨床、病理表型,因此目前NM的分類傾向于依據變異基因進行分類。盡管如此,某些變異基因可以有相似或共同的臨床/肌群受累表現,依據臨床表現,可指導基因測序方向,如ACTA-1、NEB、KLHLF40突變的NM可出現極嚴重的新生兒肌張力低下,表現為松軟兒的特征,包括胎兒運動不能或運動功能減退、關節攣縮/骨折、呼吸衰竭,并在出生時吞咽/喂養困難[6];NEB、TPM3、TPM2突變的NM,臨床表型相似,輕中度受累的患者表現出明顯的頸部肌群無力和趾屈無力,可出現足下垂和弓形足。NEB-NM在輕癥患者中可僅有小腿脛骨前肌受累,重癥患者下肢肌肉彌漫性受累[7],頭顱MRI顯示選擇性侵犯翼外肌,并影響咀嚼肌和舌肌[8]。TPM2-NM主要影響咀嚼肌(顳)及下肢遠端比目魚肌與遠端伸、屈肌,而大腿肌群無明顯受累[9]。
2 骨骼肌病理活檢
該組肌病無論是哪種致病基因所致,其特征性表現均為骨骼肌活檢于肌細胞胞漿中可見高密度的桿狀體(MGT染色觀察最為典型——紅染桿狀體)。桿狀結構的數量可隨年齡的增長而增多,但其與疾病的嚴重程度無關。不同部位的肌肉組織及不同肌纖維中桿狀體數目不盡相同,故有些NM患者沒有在桿狀體聚集的部位取材可能漏診,需行二次活檢才能明確診斷[10]。桿狀體通常出現在胞漿中且多位于肌膜下,極少病例(如ACTA1-NM)可于細胞核中觀察到桿狀體。免疫組化示桿狀體與Z線具有相同的蛋白質,α-輔肌動蛋白、肌動蛋白、肌鈣蛋白T、肌原肌球蛋白和結蛋白染色陽性[11-13]。電鏡下桿狀體呈桿狀或卵圓狀,與肌纖維長軸平行,由于桿狀體與Z線有相同的晶格結構和蛋白質,推測某些桿狀體可能來源于Z線[14]。帽狀肌病曾作為一類獨立肌病,后來研究[15-17]發現,帽狀體表現為肌膜下邊界清晰的帽狀物,出現于4%到幾乎100%肌纖維中,MGT染色為紫色偏藍或綠色,HE染色為嗜酸性,NADH-TR染色為暗藍色,ATPase染色減低,電鏡下與桿狀體結構一致,免疫組化與NM一致,故目前帽狀肌病已歸類于NM。與其他類型NM不同的是帽狀體累及肌纖維的多少與疾病的嚴重程度和患者年齡相關。軸空結構[18-19]可以與桿狀體同時存在,桿狀體聚集區域缺乏線粒體,因此該區域氧化酶染色示缺如呈軸空結構。這點也與軸空性肌病的軸空形成相鑒別,軸空性肌病的軸空結構是由于肌原纖維斷裂、排列紊亂而缺少線粒體。桿狀體并非NM特有,亦可出現正常眼肌、老年人肌肉[20]及少數遺傳性(線粒體腦肌病、強直性肌營養不良、Hodgkin病等)或獲得性神經肌肉病(皮肌炎、HIV感染等[21])。
與其他CMs一樣,NM可見肌纖維直徑大小不等,Ⅰ型肌纖維萎縮/發育不良,在肌纖維類型上呈Ⅰ型肌纖維優勢/單一Ⅰ型肌纖維。無肌纖維變性、壞死及再生。
3 分子生物學研究及發病機制
目前已發現10種基因變異(表1)可引起NM,其臨床表現具有相似或者相同的癥狀。TPM3、NEB、ACTA1、TPM2、TNNT1、CFL2編碼肌節細肌絲蛋白或相關蛋白,KBTBD13、KLHL40 (KBTBD5)和KLHL41 (KBTBD10)涉及到泛素-蛋白酶體通路(UPP),編碼小于5/6結構域的KLHL蛋白,該蛋白屬于標準KLHL蛋白家族(包括BTB、BACK、5/6 Kelch結構域)中的一種,又被歸類于KBTBD家族[19,22-29]。
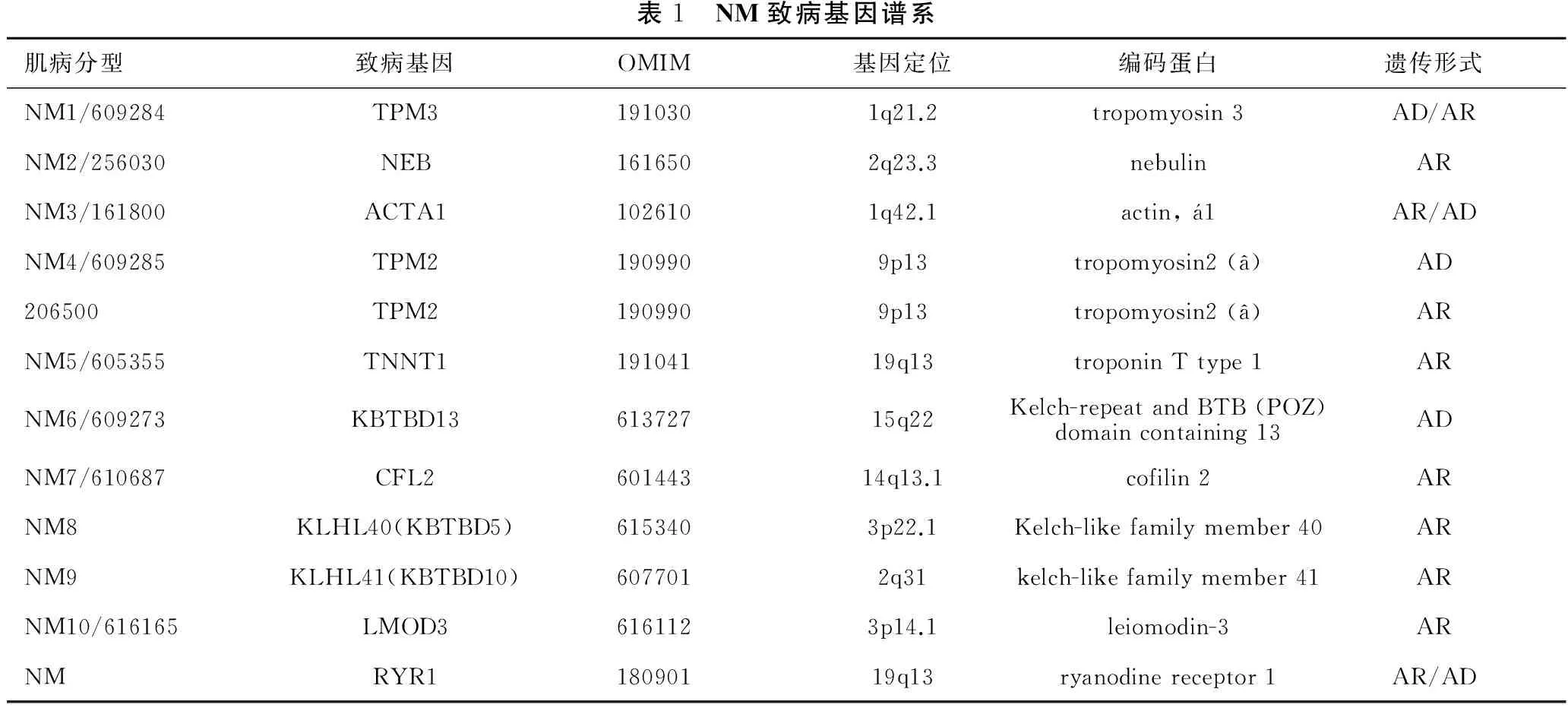
表1 NM致病基因譜系肌病分型致病基因OMIM基因定位編碼蛋白遺傳形式NM1/609284TPM31910301q21.2tropomyosin3AD/ARNM2/256030NEB1616502q23.3nebulinARNM3/161800ACTA11026101q42.1actin,1AR/ADNM4/609285TPM21909909p13tropomyosin2(a)AD206500TPM21909909p13tropomyosin2(a)ARNM5/605355TNNT119104119q13troponinTtype1ARNM6/609273KBTBD1361372715q22Kelch-repeatandBTB(POZ)domaincontaining13ADNM7/610687CFL260144314q13.1cofilin2ARNM8KLHL40(KBTBD5)6153403p22.1Kelch-likefamilymember40ARNM9KLHL41(KBTBD10)6077012q31kelch-likefamilymember41ARNM10/616165LMOD36161123p14.1leiomodin-3ARNMRYR118090119q13ryanodinereceptor1AR/AD
NEB變異是NM最常見原因,該基因擁有183個外顯子,編碼700 kDa的伴肌動蛋白。該蛋白為人類肌節中最大的蛋白之一,是細肌絲的組成部分,是肌動蛋白結合蛋白,具有高度重復序列結構,重復序列結構可以結合肌動蛋白。目前發現140個NEB突變,均呈AR遺傳且多為復合雜合突變,未發現常見的突變或熱點突變,大部分為移碼突變及無義突變,可見錯義突變、剪切突變。由于NEB巨大,以往的DNA測序使用傳統的Sanger法,僅局限于測序伴肌動蛋白的C-末端(因該端與Z線錨定),如今下一代測序技術(NGS)為NEB變異的診斷帶來了極大便利。如Scoto等[24]使用NGS報道NEB基因55號外顯子4 bp重復的移碼突變c.24372_24375dup (P.Val8126fs)。NEB還存在一些特殊突變,如1例德系猶太人于NEB基因的55號外顯子及54、55號內含子部分區域被發現2502 bp缺失突變[30],為NEB分子生物學診斷帶來了挑戰。
ACTA1是第二常見的致病基因,編碼骨骼肌a-肌動蛋白,該蛋白存在于Ⅰ型和Ⅱ型肌纖維中,約有25%的患者存在ACTA1基因突變[21]。目前已發現200余種ACTA突變,主要為錯義突變,90%為AD遺傳,10%為AR遺傳。其中,約50%的患者表現為先天性NM嚴重型。ACTA1也是唯一一個與核內桿狀體相關的基因,該基因變異亦引起斑馬體肌病,也被認為是NM的一種,目前世界上僅報道了2例,骨骼肌病理不僅發現了斑馬體,還顯示了桿狀體。
TPM2和TPM3分別編碼β-原肌球蛋白和慢型α-原肌球蛋白,遺傳方式為AR/AD,占5%以下的NM。迄今為止,已報道TPM2基因錯義突變導致的NM:如Q147P[27]、E41K[9]、E117K[31];缺失突變:p.E138del[32]、p.E139del[9-10]。TPM3已報道為AD遺傳的錯義突變及AR遺傳的1 bp的缺失突變。由于慢型α-原肌球蛋白僅存在于慢肌肌纖維中(Ⅰ型肌纖維),當肌細胞桿狀體僅存在于Ⅰ型肌纖維時,可優先考慮TPM3變異。TNNT1和CFL2突變引起NM,目前報道的均為AR遺傳。迄今為止,TNN1僅報道了1例無義突變,CFL2報道了幾例純合錯義突變及缺失突變[19,23]。CFL2編碼肌肉的肌動蛋白結合蛋白絲切因子-2,絲切因子-2缺乏可導致肌動蛋白解聚作用減少,導致其在肌纖維中的聚集形成桿狀體[28]。KBTBD13發現了3個AD遺傳的錯義突變;KLHL40、KLHL41 和LMOD3均為AR遺傳。目前報道了純合/復合雜合突變,其中KLHL41移碼突變可以導致嚴重的臨床表現,新生兒即死亡;錯義突變損害運動功能,患者可以存活至兒童晚期或成年早期[33]。
RYR1導致的單純的NM非常少見,目前僅報道1例[34],該研究使用NGS發現1例2歲極嚴重NM患者的RYR1第33號外顯子c.4718C>T (p.1573Pro>Leu)及47號外顯子c.7585G>A (p.2529Asp>Asn) 復合雜合突變。RYR1變異報道較多的是軸空/桿狀體肌病[18, 34-35]。軸空/桿狀體肌病在NM中非常少見,已報道致病基因除了最多見的RYR1外還有NEB[22]和ACTA1[36]。帽狀體肌病目前已知的致病基因為ACTA1[14]、TPM2[9, 11]、TPM3[37-38]。
4 總結
隨著分子生物學技術的發展與應用,越來越多的NM病例被報道,人們認識了更多的NM的臨床、病理及遺傳學特征,因此,“桿狀體”不能再作為診斷本組肌病的金標準,而是必須結合分子生物學診斷。同時,分子生物學研究的進展也推動了人們對該病發病機制的探索,隨著動物模型的建立,NM的發病機制研究以及基因治療將成為新的研究熱點。
[1] Colombo I, Scoto M, Manzur AY, et al. Neurology, 2015, 84: 28.
[2] North KN, Wang CH, Clarke N, et al. NMD, 2014, 24: 97.
[3] Lehtokari VL, Pelin K, Herczegfalvi A, et al. NMD, 2011, 21: 556.
[4] Wallgren-Pettersson C, Laing NG. NMD, 2000, 10: 299.
[5] Ryan MM, Schnell C, Strickland CD, et al. Ann Neurol, 2001, 50: 312.
[6] Ravenscroft G, Miyatake S, Lehtokari VL, et al. Am J Hum Genet, 2013, 93: 6.
[7] Jungbluth H, Sewry CA, Counsell S, et al. NMD, 2004, 14: 779.
[8] Quijano-Roy S, Avila-Smirnow D, Carlier RY. NMD, 2012, 22: 68.
[9] Tajsharghi HOM, Lindberg C, Oldfors A. Arch Neurol, 2007, 64: 1334.
[10] 尹西, 蒲傳強, 黃旭升, 等.中華神經科雜志, 2013, 46: 676.
[11] Clarke NF, Domazetovska A, Waddell L, et al. NMD, 2009, 19: 348.
[12] de Paula AM, Franques J, Fernandez C, et al. NMD, 2009, 19: 685.
[13] Ohlsson M, Fidzianska A, Tajsharghi H, et al. Neurology, 2009, 72: 1961.
[14] Hung RM, Yoon G, Hawkins CE, et al. NMD, 2010, 20: 238.
[15] Waddell LB, Kreissl M, Kornberg A, et al. NMD, 2010, 20: 464.
[16] Malfatti E, Schaeffer U, Chapon F, et al. NMD, 2013, 23: 992.
[17] Ohlsson M, Quijano-Roy S, Darin N, et al. Neurology, 2008, 71: 1896.
[18] Scacheri P, Hoffman E, Fratkin J, et al. Neurology, 2000, 55: 1689.
[19] Hernandez-Lain A, Husson I, Monnier N, et al. Eur J Med Genet, 2011, 54: 29.
[20] Wallgren-Pettersson C, Sewry CA, Nowak KJ, et al. Semin Pediatr Neurol, 2011, 18: 230.
[21] de Sanctis JT, Cumbo-Nacheli G, Dobbie D, et al. AIDS Read, 2008, 18: 90.
[22] Romero NB, Lehtokari VL, Quijano-Roy S, et al. Neurology, 2009, 73: 1159.
[23] Wattanasirichaigoon D, Swoboda K, Takada F, et al. Neurology, 2002, 59: 613.
[24] Scoto M, Cullup T, Cirak S, et al. Euro J Hum Genet, 2013, 21: 1249.
[25] Ravenscroft G, Wilmshurst JM, Pillay K, et al. NMD, 2011, 21: 31.
[26] Donner K, Ollikainen M, Ridanp?? M, et al. NMD, 2002, 12: 151.
[27] Johnston JJ, Kelley RI, Crawford TO, et al. Am J Hum Genet, 2000, 67: 814.
[28] Agrawal PB, Greenleaf RS, Tomczak KK, et al. Am J Hum Genet, 2007, 80: 162.
[29] Dhanoa BS, Cogliati T, Satish AG, et al. Hum Genet, 2013, 7: 13.
[30] Anderson SL, Ekstein J, Donnelly MC, et al. Human Genet, 2004, 115: 185.
[31] Karpicheva OE, Robinson P, Piers A, et al. Arch Biochem Biophys, 2013, 536: 25.
[32] Tasca G, Fattori F, Ricci E, et al. Acta Neuropathol, 2013, 125: 169.
[33] Gupta-Vandana A, Ravenscroft G, Shaheen R, et al. Am J Hum Genet, 2013, 93: 1108.
[34] Kondo E, Nishimura T, Kosho T, et al. Am J Hum Genet, 2012, 158: 772.
[35] Davis M, Haan E, Jungbluth H, et al. NMD, 2003, 13: 151.
[36] Jungblutha H, Browna SC, Nowak KJ, et al. NMD, 2001, 11: 35.
[37] Schreckenbach T, Schroder JM, Voit T, et al. NMD, 2014, 24: 117.
[38] Fidzianska A, Madej-Pilarczyk A, Hausmanowa-Petrusewicz I. Clin Neuropathol, 2014, 33: 61.
R746
A
1004-1648(2017)05-0383-03
國家自然科學基金青年基金(81300977);國家自然科學基金面上項目(81671113)
210008南京大學醫學院附屬鼓樓醫院神經內科(趙燕,劉卓,常蕾蕾,盧正娟,吳家勇,徐運),病理科(牛豐南)
徐運
2016-12-10
2016-12-20)
