額葉p-mTOR異常表達與外傷性癲癇發生的相關研究
2018-05-07 02:05:42張艷麗吐爾遜沙比爾
中國康復 2018年2期
關鍵詞:癲癇
張艷麗,吐爾遜·沙比爾
外傷性癲癇(Posttraumatic epilepsy,PTE)是繼發于創傷性腦損傷(Traumatic brain injury,TBI)后的發作性功能異常。PTE是腦外傷的常見后遺癥,在TBI患者中有著較高的發病率和死亡率[1],同時也是臨床上難治性癲癇的常見原因。哺乳動物雷帕霉素靶蛋白(mammalian target of rapamycin,p-mTOR)是一種絲氨酸/蘇氨酸蛋白激酶,具有復雜的生物學效應,越來越多的證據表明mTOR異常激活可誘導PTE的發生,并在TBI患者及PTE動物模型中mTOR通過磷酸化被激活,活化的mTOR可通過磷酸化下游多種效應分子產生多重病理生理改變從而引起癲癇的進一步發展[2]。既往研究發現人與鼠的mTOR序列一致性高達98%[3]。目前在其他病因引起的癲癇中發現mTOR通路的異常激活,但mTOR信號通路在PTE中的激活情況及與PTE的關系目前還不明確[4-5]。目前PTE的動物模型有鐵離子模型、液壓打擊模型、自由落體模型、控制性皮質撞擊模型等,由于鐵離子模型接近人類腦外傷后出血的病理及臨床改變,成功率高,便于觀察及干預性的研究,同時額葉皮質定位操作簡單,故本實驗通過常用的右額葉注射FeCl2法建立PTE大鼠模型[6],觀察額葉皮質mTOR通路相關蛋白p-mTOR(Ser2448)的動態表達,探討其在PTE中的發病作用,為PTE的防治提供理論基礎。
1 材料與方法
1.1 研究對象 健康成年雄性SD大鼠78只,體質量180~220g,分為正常對照組(A組,n=6),生理鹽水對照組(B組,n=36)和癲癇模型組(C組,n=36)。
1.2 方法 ①PTE模型的制作:大鼠以10%水合氯醛(350mg/kg)腹腔麻醉,術區備皮后固定于大鼠立體定向儀頭架上,消毒,暴露顱骨,生理鹽水組和癲癇模型組大鼠右額葉皮質的鉆孔坐標為:前囟前2.0mm,中線旁3.0mm,硬膜下2.0mm,然后微量進樣器緩慢進針至靶點,分別向靶點緩慢注射10uL的生理鹽水或FeCl2(0.1mol/L)[6],FeCl2粉劑由武漢萊康生物提供。速率均為1μL/min,注射后留針5min。麻醉清醒后進行行為學檢測,根據 Racine分級判斷模型是否成功,0級:無任何癲癇發作;I級:凝視發作;II級:出現規律性點頭或濕狗樣抖動,伴有或不伴有面部抽動記;III級:一側前肢震顫 ;IV級:站立、雙前肢震顫及持續性點頭;V級:前肢震顫加重,失去平衡跌倒而出現全身性強直-陣攣性發作;VI級:發作衰竭導致死亡,出現 4 級或以上發作行為為制模成功[7]。②動物處理:分別于制模后1h、24h、1周、2周、4周,先分別從A、B、C組各取6只,腹腔麻醉(以100g/L水合氯醛300g/kg腹腔麻醉)后,夾閉腹主動脈脈和下腔靜脈,經升主動脈插管,灌注生理鹽水200ml,再灌注含4%多聚甲醛的0.1mol/L PBS(pH7.4,4℃)300ml,緩慢灌注至肢體完全僵硬、斷頭完整取腦,矢狀切片,福爾馬林固定,石蠟包埋,石蠟包埋后切片厚3μm。 二甲苯脫蠟3次,每次2min,然后切片常規脫蠟、水化(環保透明脫蠟液30min→無水乙醇5s→95%乙醇10s→80%乙醇5s→自來水水洗3min→蒸餾水3min)。
1.3 檢測指標 ①HE染色:將水化后的切片放入蘇木精染色1~2min,蒸餾水沖洗數秒,1%鹽酸酒精(70%酒精99ml+濃鹽酸1ml)沖洗10~30s,1%氨水酒精(70%酒精99ml+濃氨水1ml)沖洗數秒至30s不等,蒸餾水沖洗3min后使用伊紅染色40s,最后蒸餾水沖洗至無色。②免疫組化染色:組織切片后60℃烘片10h后二甲苯脫蠟2次,梯度乙醇脫水,蒸餾水沖洗凈,PBS漂洗;將玻片浸入3%過氧化氫溶液中,PBS漂洗;滴加一抗,兔抗人p-mTOR(Ser2448)單克隆抗體由美國Cell Signaling Technology公司提供,4℃孵育過夜,室溫中放置后PBS漂洗,滴加聚合物輔助劑,室溫孵育,PBS液漂洗;滴加二抗,抗兔通用型免疫組化試劑盒,購于丹麥Dako公司。室溫孵育,PBS漂洗;滴加DAB溶液顯色,蒸餾水終止反應,棕黃色為染色陽性;蘇木素復染后自來水沖洗,依次置于梯度乙醇脫水、二甲苯透明;中性樹膠封片,觀察結果。半定量分析p-mTOR(Ser2448)在額葉中的平均光密度(Mean Optical Density,MOD)值。

2 結果
2.1 HE染色結果 A組(圖1a、b):組織結構清晰,額葉的皮層未見神經元變性。B組(圖1c、d):額葉可見螺旋狀神經元纖維,胞漿變性、細胞腫脹不明顯。C組(圖1e、f):額葉皮質可見鏤空或網狀病灶,細胞層次紊亂,神經元嚴重變性,細胞核固縮、深染,細胞核仁消失,膠質細胞明顯增生,可見噬神經現象。

圖1 額葉皮層神經元HE染色結果(×400)
圖1a、c、e中的方框代表低倍鏡下隨機選取的視野,圖1b、d、f分別為隨機視野的高倍鏡圖像,箭頭代表變性的神經元細胞
2.2 免疫組化結果 免疫組化定位p-mTOR(Ser2448)在額葉中主要表達在神經元的細胞質中(p-mTOR抗原與抗體結合后顯示為棕色),細胞核及細胞外未見表達。圖2顯示1周后 A組(圖a、b)額葉p-mTOR(Ser2448)表達的免疫組織化學染色,神經元胞質呈淺棕色,提示弱陽性表達; B組(圖c、d)細胞染色程度較A組加深,p-mTOR(Ser2448)表達陽性細胞數增加;C組(圖e、f)可見細胞結構紊亂,胞質呈明顯的棕黃色,提示該蛋白在細胞質中呈強陽性表達。C組和B組比較,1h表達開始升高(t=-1.435,P=0.182),但差異并不顯著,1周表達高峰(t=-4.073,P=0.002),2周降低(t=-2.614,P=0.026),4周時再次升高(t=-2.506,P=0.031),差異具有統計學意義(均P<0.05 ),見表1。

圖2額葉皮層神經元免疫組化結果a 、c 、e(×200),b、 d 、f(×400)
圖2a、c、e中的方框代表低倍鏡下隨機選取的視野,圖2b、d、f分別為隨機視野的高倍鏡圖像,圖2d,f箭頭所指分別代表p-mTOR弱陽性表達和強陽性表達的神經元
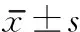

時間B組C組tP1h2.108±0.4002.318±0.498-1.4350.18224h2.427±0.4952.778±0.515-2.9690.0141周2.603±0.5723.133±0.466-4.0730.0022周2.247±0.4882.543±0.505-2.6140.0264周2.345±0.4182.672±0.533-2.5060.031
3 討論
根據目前已有的研究結果,mTOR信號通路參與了細胞凋亡、吞噬、膠質增生、突觸可塑性、神經遞質受體、離子通道、軸突芽生等癲癇多種分子水平、細胞水平的病理改變,上游信號通路PI3K/Akt磷酸化后可激活mTOR,mTOR是一種相對保守的非典型絲/蘇氨酸蛋白激酶,具有復雜的下游效應,其中與PTE的發生有著重要聯系的是p-mTOR C1介導的下游信號通路,包括核糖體S6通路和4EBP1通路,S6屬于AGC激酶家族的絲氨酸/蘇氨酸激,S6經p-mTOR C1激活后,磷酸化的S6k再將S6磷酸化,這樣使得含嘧啶基序序列mRNA的翻譯功能增強;4EBPl也是p-mTOR C1的另外一個靶點,當mTOR信號通路激活時,4EBPl會因被p-mTOR C1磷酸化而失活,導致4EBPl從真核細胞翻譯因子4E(eIF-4E)釋放出來,從而降低4EBPl對蛋白翻譯的抑制作用,最終導致額葉神經元興奮性增高,異常細胞同步放電,最終導致癲癇的發生[8-9]。本課題組通過向大鼠右側額葉皮質內注射鐵離子制造PTE動物模型,該研究中額葉p-mTOR染色MOD最大的細胞主要分布于顆粒細胞層的胞質,同時分子層及多形細胞層也可見散在陽性細胞分布。實驗結果顯示,p-mTOR在正常大鼠額葉少量表達;本研究結果提示PTE動物模型中存在p-mTOR的表達增高,這與2009年Codeluppi[10]在PTE患者反應性星形膠質細胞中發現mTOR激活的結果相似,考慮可能是由于PTE引起的一系列氧化應激反應導致額葉顆粒細胞進一步反應性增殖,以及氧化應激后導致已經成熟的神經元或膠質細胞增殖分化[11],所以1周時MOD值最高;C組在2周時,p-mTOR的表達下降并接近正常水平,與B組該時間的比較差異具有統計學意義,分析原因可能與1周時產生的神經前體細胞分化為比較成熟的膠質細胞及損傷區神經元、膠質細胞壞死有關,故表達下降,但Sha等[12]研究顯示p-mTOR的表達增高仍可持續到2周,原因可能與不同研究者采用不同的動物模型有關,具體機制還有待于進一步深入研究。在本研究中C組4周組p-mTOR表達再次升高,可能與發作性的癲癇發作導致的應激反應引起額葉顆粒細胞進一步反應增殖等因素有關[13]。
通過在大鼠額葉注射FeCl2引起大鼠PTE,并在大鼠額葉齒狀回觀察到反應性增殖的顆粒細胞,其增殖和分化可能與氧化應激反應導致mTOR通路的異常活化以及發作性癲癇的發生有關聯,最終導致神經膠質細胞的過度增生,神經元數量減少、功能異常,海馬結構紊亂[14-16];同時在損傷灶周圍皮質觀察到反應性增生的錐體神經元及膠質細胞,其增殖和分化也可能與FeCl2氧化應激刺激導致mTOR通路的異常活化有關聯,導致皮質結構的紊亂,從來誘發癲癇發作[17-18],后期陣發性癲癇的刺激與mTOR通路的激活互為因果關系,相互促進進一步誘發慢性癲癇。同時我們觀察到mTOR信號通路的異常激活在1周時最高,2周時降低,4周時又再次升高,和PTE大鼠的平均增加癲癇發作次數改變趨勢相似,這種mTOR通路的異常激活可能是導致晚期PTE發作的重要證據[19-20]。然而,反應性增生的神經元及膠質細胞與mTOR的異常激活還需進一步的觀察,是否可以使用mTOR通路的抑制劑RPN抑制mTOR的異常表達來治療PTE還需進一步研究。本研究雖然發現PTE的發病機制可能與mTOR信號通路上額葉區p-mTOR(Ser2448)的表達異常增加有關,但并未涉及顳葉及海馬區相關蛋白的研究,故還需要進一步深入探索。
【參考文獻】
單因素分析結果表明,導致結腸癌合并腸梗阻患者術后傷口感染顯著性相關的因素有體重指數、合并糖尿病、術前化療、手術時間、術前低蛋白、術后引流管放置時間等(P<0.05),見表3。
[1] Frey LC. Epidemiology of posttraumatic epilepsy: a critical review[J]. Epilepsia, 2003, 44(10): 11-17.
[2] Cho CH. Frontier of epilepsy research-mTOR signaling pathway[J]. Experimental & molecular medicine, 2011, 43(5): 231-274.
[3] G?del M, Hartleben B, Herbach N, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice[J]. The Journal of clinical investigation, 2011, 121(6): 2197-2209.
[4] Shima A, Nitta N, Suzuki F, et al. Activation of mTOR signaling pathway is secondary to neuronal excitability in a mouse model of mesio‐temporal lobe epilepsy[J]. European Journal of Neuroscience, 2015, 41(7): 976-988.
[5] Bockaert J, Marin P. mTOR in brain physiology and pathologies[J]. Physiological reviews, 2015, 95(4): 1157-1187.
[6] 林元相, 徐如祥, 姜曉丹,等. 皮層注射氯化亞鐵建立外傷性癲癇動物模型[J]. 中華神經醫學雜志, 2006, 5(4):372-377.
[7] Lüttjohann A, Fabene PF, van Luijtelaar G. A revised Racine's scale for PTZ-induced seizures in rats[J]. Physiology & behavior, 2009, 98(5): 579-586.
[8] Tee AR, Sampson JR, Pal DK, et al. The role of mTOR signalling in neurogenesis, insights from tuberous sclerosis complex[C]//Seminars in cell & developmental biology. Academic Press, 2016, 52(1): 12-20.
[9] Pitk?nen A, Lukasiuk K. Mechanisms of epileptogenesis and potential treatment targets[J]. The Lancet Neurology, 2011, 10(2): 173-186.
[10] Codeluppi S, Svensson C I, Hefferan M P, et al. The Rheb-mTOR pathway is upregulated in reactive astrocytes of the injured spinal cord.[J]. Journal of Neuroscience the Official Journal of the Society for Neuroscience, 2009, 29(4):1093-1104.
[11] Althaus A L, Sagher O, Parent J M, et al. Intrinsic neurophysiological properties of hilar ectopic and normotopic dentate granule cells in human temporal lobe epilepsy and a rat model[J]. Journal of neurophysiology, 2015, 113(4): 1184-1194.
[12] Sha L Z, Xing X L, Zhang D, et al. Mapping the spatio-temporal pattern of the mammalian target of rapamycin (mTOR) activation in temporal lobe epilepsy.[J]. Plos One, 2012, 7(6):391-412.
[13] Romá-Mateo C, Aguado C, García-Giménez J L, et al. Oxidative stress, a new hallmark in the pathophysiology of Lafora progressive myoclonus epilepsy[J]. Free Radical Biology and Medicine, 2015, 88 (1): 30-41.
[14] Ostendorf A P, Wong M. mTOR Inhibition in Epilepsy: Rationale and Clinical Perspectives[J]. CNS drugs, 2015, 29(2): 91-99.
[15] Binder D K, Steinh?user C. Functional changes in astroglial cells in epilepsy[J]. Glia, 2006, 54(5): 358-368.
[16] Lipton J O, Sahin M. The neurology of mTOR[J]. Neuron, 2014, 84(2): 275-291.
[17] Kharatishvili I, Pitk?nen A. Posttraumatic epilepsy[J]. Current opinion in neurology, 2010, 23(2): 183-188.
[18] Berdichevsky Y, Dryer A M, Saponjian Y, et al. PI3K-Akt signaling activates mTOR-mediated epileptogenesis in organotypic hippocampal culture model of post-traumatic epilepsy[J]. The Journal of Neuroscience, 2013, 33(21): 9056-9067.
[19] Russo E, Citraro R, Constanti A, et al. The mTOR signaling pathway in the brain: focus on epilepsy and epileptogenesis[J]. Molecular neurobiology, 2012, 46(3): 662-681.
[20] Meng X F, Yu J T, Song J H, et al. Role of the mTOR signaling pathway in epilepsy[J]. Journal of the neurological sciences, 2013, 332(1): 4-15.
猜你喜歡
中國民間療法(2021年5期)2021-06-09 09:21:04
中華養生保健(2020年2期)2020-11-16 00:49:00
解放軍醫學院學報(2020年12期)2020-03-29 05:11:46
中成藥(2017年6期)2017-06-13 07:30:35
飲食科學(2017年5期)2017-05-20 17:11:53
臨床醫藥文獻雜志(電子版)(2017年11期)2017-05-17 04:48:10
安徽醫科大學學報(2015年9期)2015-12-16 11:09:44
中國當代醫藥(2015年7期)2015-03-01 02:01:13
西南軍醫(2015年4期)2015-01-23 01:19:30
西部中醫藥(2014年6期)2014-03-11 16:07:47
