甘油尿素法合成甘油碳酸酯的催化劑研究進展
2018-06-05 05:38:54王洪平王澤溟張世龍王家喜
石油化工 2018年5期
關鍵詞:催化劑
王洪平,梁 爽,王澤溟,張世龍,王家喜
(河北工業大學 化工學院,天津 300130)
生物柴油工業的快速發展致使大量的副產物甘油嚴重過剩,并影響了生物柴油的經濟化生產。因此,開發副產物甘油的新工業用途至關重要[1]。甘油碳酸酯(GC)是甘油的高附加值衍生產品。GC具有低易燃性、低毒性、高沸點和良好的生物降解性,成為各種應用中非常有吸引力的化學品,廣泛用于質子溶劑、聚合物化學中間體和鋰電池電解質[2]等方面。目前,由甘油合成GC的方法主要包括甘油光氣法、甘油CO2法、甘油碳酸二甲酯法及甘油與尿素法等[3-5]。由于光氣具高毒性、甘油CO2超臨界法技術實施投資大以及甘油碳酸烷基酯法的經濟效益差等問題,所以由廉價的甘油與尿素反應合成高附加值產品GC一直是人們研究的熱點。甘油與尿素反應過程中產生的副產物NH3易于除去,NH3可與CO2反應轉化為尿素,整個過程形成了綠色化學循環[6]。甘油與尿素的反應需要催化劑的催化,因此高效、高選擇性催化劑也成為了人們的研究重點。盡管尿素醇解法合成有機碳酸酯[7]、GC的其他合成方法已有文獻概述[3-4,8],但甘油與尿素反應合成GC的研究還未見文獻總結。
本文從均相與多相催化兩個角度分別綜述了不同種類甘油尿素法合成的GC催化劑的催化性能,介紹了金屬鹽、金屬氧化物、離子液體(ILs)等不同種類催化劑在甘油尿素法合成GC中的應用,提出了可能的反應機理,為進一步優化催化劑的結構提供了有益的幫助。
1 均相催化劑
1.1 金屬鹽類催化劑
2012年,本課題組[9]探討了ZnSO4水合物的活化條件對催化活性的影響。實驗結果表明,在390 ℃下煅燒3 h的ZnSO4催化效果最好,GC產率達到88.3%。催化劑中Lewis 酸性的Zn2+與尿素分子中具有Lewis堿性的羰基氧配位,SO42-上Lewis堿性的氧與甘油上的羥基氫相互作用,從而同時活化兩種原料發生催化反應。為了解決產品分離問題,河北工業大學[10]提出了首先將甘油與尿素在鋅鹽(Zn(OAc)2和ZnSO4等)催化下合成GC,再以K2CO3為催化劑催化反應體系中剩余的甘油與碳酸二甲酯反應,降低甘油的含量,最后得到甘油含量僅為0.6%的GC,產率高達97.0%。
Park等[11]也探究了一系列鋅基催化劑(ZnBr2,ZnI2等)的催化活性。相比之下,ZnCl2顯示出良好的催化活性。ZnCl2在反應過程中不僅可完全溶于反應體系中,而且Cl-也具有一定的催化活性。
2015年,Wang等[12]利用了不同的鑭化合物(La(NO3)3和LaCl3等)催化尿素與甘油合成GC。其中使用LaCl3作催化劑時催化效果最佳。在150 ℃下反應3 h,甘油轉化率為95.4%,GC選擇性為99.7%。
1.2 雜多酸類催化劑
近年來,利用雜多酸獨特的酸性特性開發了用金屬離子交換的鎢磷酸固體酸催化劑。其中,經不同金屬離子交換制備的改性鎢磷酸催化劑對甘油羰基化合成GC的反應具有良好的催化性能。Kumar等[13-14]制備了Sm3+和Zn2+交換的鎢磷酸催化劑(SmxTPA和ZnxTPA)。研究發現這種催化劑的活性與其酸度有關,而催化劑的酸度與Sm或Zn的含量相關。其中,Sm0.66TPA與Zn1TPA催化劑的酸度最大,催化活性最高。Babu等[15]探究了由鉭交換的鎢磷酸催化劑(TaxTPA)。Ta0.4TPA催化甘油與尿素反應獲得約71.0%的甘油轉化率和約100%的GC選擇性。NH3-TPD分析結果表明,該催化劑具有較高的酸性,所以該催化劑的高活性源于強酸性。
1.3 其他催化劑
Chen等[16]考察了不同酸性、堿性和中性的ILs的催化性能。由于陽離子和陰離子的協同效應,中性ILs在反應中具有較高的催化活性。推測具有正電荷的陽離子會活化尿素,帶負電荷的陰離子會活化甘油。
Turney等[17]分別制備了Zn和Co與甘油形成甘油金屬絡合物,其中Zn的甘油金屬絡合物作均相催化劑時顯示出較好的催化性能,甘油轉化率高達98.0%,GC選擇性高達85.0%。
2 非均相催化劑
2.1 金屬氧化物類催化劑
在甘油尿素羰基化合成GC的研究中,金屬氧化物(二元或混合金屬氧化物)催化劑是最大的非均相催化劑體系。值得注意的是,具有Lewis酸和堿基位點組合的催化劑表現出更好的催化性能。
Marcos等[18-19]制備Co3O4/ZnO復合催化劑催化甘油和尿素的反應。實驗結果表明,在140 ℃下反應4 h,甘油轉化率可以達到69.0%,GC選擇性高達97.0%。此外,通過ATR-FTIR監測了催化合成GC的反應過程,解釋了相應的反應機理。
Wang等[20]首次使用氧化鑭作固體堿催化劑,并考察了煅燒溫度的影響。實驗結果表明,在600 ℃下煅燒后的La2O3在反應過程中表現出良好的催化性能,即催化劑的催化速率達到最佳值(1 506 mmol/(g·h))。為了進一步了解鑭基催化劑在該反應中的催化性能,Zhang等[21]通過溶膠-凝膠法制備鑭基混合氧化物La2Cu0.5Fe0.5O4,該復合物在950 ℃下煅燒后顯示出較高的催化活性。催化劑回收利用6次后仍然保持較好的催化活性。
Baek等[22]制備了混合氧化物ZnAl2O4。在500℃下煅燒后的ZnAl2O4可達74.4%的甘油轉化率及98.5%的GC選擇性。通過調節Zn/Al摩爾比以及催化劑的制備條件,考察了復合混合氧化物的催化活性與結構的關系。實驗結果表明,當Zn/Al摩爾比為7∶3時,甘油轉化率為 82.7%,GC選擇性高達 99.5%[23-24]。在此基礎上,Phu 等[25]將活性 Zn/Al混合氧化物分散到工業廢料-活性紅泥(ARM)中考察催化性能。FTIR分析結果表明,ARM具有第1步的催化活性,而Zn/Al氧化物在所有反應步驟中都顯示出催化活性,反應后甘油轉化率為69.0%,GC選擇性達84.2%。
Jagadeeswaraiah等[26]討論了錫鎢混合氧化物中Sn/W摩爾比對甘油與尿素羰基化反應的影響。當Sn/W摩爾比為2∶1時,在500 ℃下活化后表現出最佳的催化活性。此外,還制備了一系列TiO2負載WO3的催化劑[27]。實驗結果表明,15%(w)WO3負載在TiO2(最佳載體)上的復合氧化物在500 ℃下煅燒后的催化性能較好。
Pandian等[28]制備了一種新型Zn-Sn復合氧化物,Zn/Sn摩爾比為2∶1的Zn-Sn復合氧化物在600 ℃下煅燒后顯示出較好的催化活性。此外,認為兩種金屬陽離子與氧的不同連接方式導致形成不同的陽離子環境,從而產生不同的活性中心。
2.2 離子液體類催化劑
Kim等[29]制備了聚苯乙烯基的Merri field肽樹脂負載的離子液體催化劑(MPR-ILs),催化甘油尿素反應時的甘油轉化率達78.3%,GC選擇性為94.4%。后來Kim等[30-31]又比較了載體(聚苯乙烯(PS)和商業二氧化硅等)與負載離子液的結構與催化活性的關系,研究發現由PS負載的含金屬的咪唑鎓鹽(PS-(Im)2ZnBr2和PS-(Im)2ZnI2)的催化活性較高,甘油轉化率分別為65.8%和71.7%,GC選擇性分別為72.3%和84.1%。催化劑易于分離回收,可循環使用。Kim等[32]又制備了以Lewis酸ZnX2與Lewis堿性的有機胺形成的雙功能酸堿催化劑((RIm)2ZnX2)。1-(2-羥乙基)咪唑/ZnCl2在140 ℃條件下催化甘油與尿素反應可達92.7%的甘油轉化率與93.4%的GC選擇性。
2.3 分子篩類催化劑
Hammond等[6]利用具有高比表面積且熱穩定性很高的新型沸石分子篩ZSM-5作為分散Au的載體制備了Au/ZSM-5催化劑。在400 ℃下煅燒后該催化劑的甘油轉化率為81.0%,GC選擇性為68.0%。催化劑循環使用10次后仍保持較好的活性。
Kondawar等[33]制備了一系列分子篩MCM-41孔內固載不同過渡金屬(Cu,Ni,Zn)的多相催化劑。其中,Zn/MCM-41催化劑的催化活性最高,甘油轉化率為75.0%,GC選擇性高達98.0%。Marakatti等[34]比較了Zn改性的不同沸石,如ZSM-5、八面沸石(FAU)等的催化活性。含0.79 mmol/g Zn的Zn-FAU對甘油與尿素反應顯示出較好的催化活性,甘油轉化率94.6%,GC選擇性高達98.0%。
Narkhede等[35]采用硅鎢酸作為高活性固體酸催化劑催化合成GC。其中,浸漬到MCM-41中的單壁硅鎢酸鹽SiW11具有較好的催化性能,甘油轉化率為75.0%,GC選擇性為77.0%。
2.4 雙金屬氫氧化物(類水滑石)類催化劑
Climent等[36]制備了不同酸堿性的水滑石固體催化劑 (HTc-Zn和HTc-Zn/Li等)。通過對比催化效果可知,具有平衡酸堿性位點的催化劑催化性能最佳。其中,HTc-Zn是具有高活性和選擇性的催化劑,若引入Li離子可以提高水滑石堿度,但GC產率大幅降低。
Sun等[37]研究發現,與添加Zn相比,添加Ga和Ni不利于Mg-Al水滑石的催化活性,這與這些金屬鹵化物的活性一致。研究結果表明,HT(Mg/Zn/Al)催化劑對于該方法顯示出高的催化活性,在反應過程中保持高穩定性。
Sandesh等[38]通過共沉淀法合成了 ZnSn(OH)6類水滑石固體催化劑。當Zn/Sn摩爾比為2∶1時,在600 ℃下煅燒后Zn-Sn復合催化劑的催化性能較好。催化劑循環使用4次,活性略有下降。
2.5 其他催化劑
Lee等[39]制備了具有季銨基團的功能化金屬有機骨架材料F-IRMOF-3(BuI)。該催化劑由親電中心Zn4O和親核中心X-組成。F-IRMOF-3(BuI)作為新型單組分多相催化劑用于無溶劑合成GC,具有較大的烷基鏈結構和更親核的陰離子的F-IRMOF-3(BuI),在GC的合成中表現出較好的活性。
隨著對工業廢棄物再利用認識的加深,Indran等[40-42]首次使用棕櫚油廠的廢鍋爐灰(主要組分為K2SiO3)作催化劑。通過考察各種煅燒溫度以及與商業K2SiO3對比,認為鍋爐灰中的K2SiO3組分是催化合成GC的有效催化劑,K+作為弱Lewis酸活化尿素的羰基,SiO32-基團作為有效的共軛堿性位點活化甘油的羥基以催化甘油與尿素的反應。同樣,Zuhaimi等[43]首次報道了將先進材料制造業的廢料石膏(CaSO4·2H2O)作為非均相催化劑催化甘油與尿素的羰基化反應。其中β-CaSO4相催化劑的催化活性較高,認為Ca2+作為路易斯酸位點與SO42-作為石膏催化劑中的共軛堿基位點是負責催化活性和選擇性的原因。
3 催化劑催化性能的對比及反應機理
均相催化劑溶解于反應體系中活化尿素及甘油分子,由于催化劑與反應物能達到分子級混合,催化活性與催化劑結構的關系可調,但催化劑結構的穩定性及催化劑的分離是科學界及工業界面臨的重大問題。相對而言,多相催化劑盡管催化劑的結構精確表征較困難,催化劑的催化性能與催化劑結構的構效關系研究還存在不足,但多相催化劑活性點相對穩定,使用壽命相對較長,易分離回收再利用及部分多相催化劑催化性能優異等優點使得多相催化劑備受工業界的關注。尿素與甘油反應中一些催化劑的催化性能歸納總結于表1。
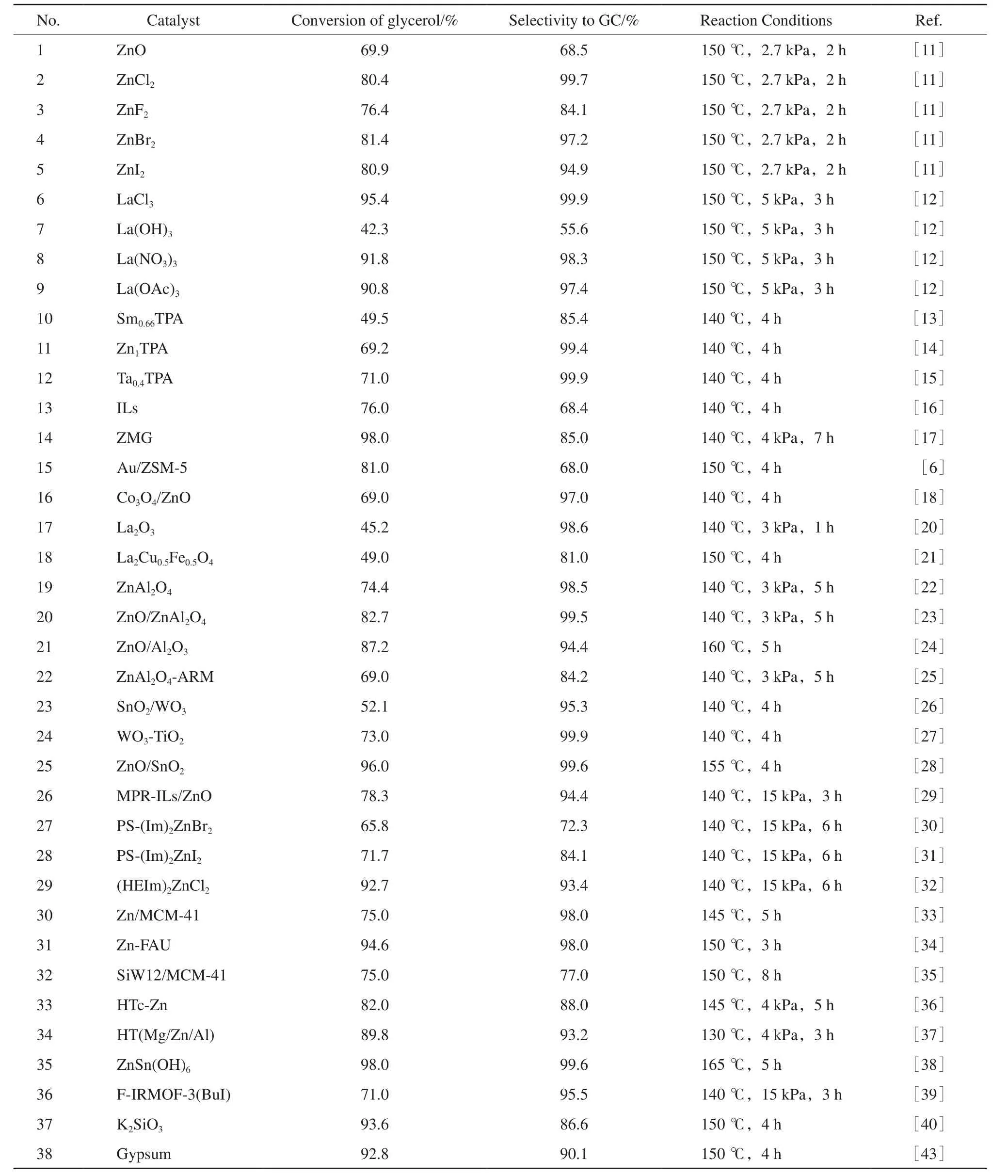
表1 甘油與尿素反應中催化劑的催化性能Table 1 Catalytic properties of catalysts in the reaction of glycerol with urea
由表1可見,在均相催化劑中,具有強Lewis酸性的鑭鹽(LaCl3,La(NO3)3,La(OAc)3)的催化活性及GC選擇性高,La離子與尿素配位,提高了尿素的反應活性,而強堿性的La(OH)3催化活性及GC的選擇性都較低。具有強Lewis酸性的鹵化鋅催化劑的高GC選擇性也進一步證明酸性催化劑有利于提高GC選擇性。相對于La(OH)3,高溫脫水后的多相催化劑La2O3的堿性下降,Lewis酸性增強,GC的選擇性得到提高。具有酸堿雙功能活性位點且酸堿活性性能匹配的催化劑是甘油羰基化合成GC反應中最高效的催化劑(No.25和No.35)。從分子反應的角度分析,尿素分子由于存在很強的分子間氫鍵,氨基由于對羰基具有吸電子的誘導效應及給電子的共軛效應而非常穩定,為了提高羰基的反應活性,催化劑必須有酸性催化活性位,酸性位點與尿素的羰基氧配位可以削弱尿素間的氫鍵強度,提高尿素與親核試劑反應的活性。甘油分子間及分子內的氫鍵也降低了甘油的親核性,催化劑的堿性位點可以使甘油轉化成甘油基氧負離子,以提高甘油親核性。
Climent等[36]基于水滑石基催化劑提出了尿素與甘油反應的Lewis酸性位點與堿性位點協同作用的反應機理。Casilda等[19]通過ATR-FTIR監測到反應過程中中間體氨基甲酸甘油酯的生成。結合文獻,得出甘油與尿素的反應機理,如圖1所示。催化劑中堿性位點活化甘油中的羥基,酸性位點活化尿素中的羰基,活化后的甘油與尿素反應,脫出氨氣,形成氨基甲酸酯及最后的GC。具有酸堿雙功能活性位點的催化劑可以同時活化尿素及甘油,協同作用使其可望達到最佳催化性能。
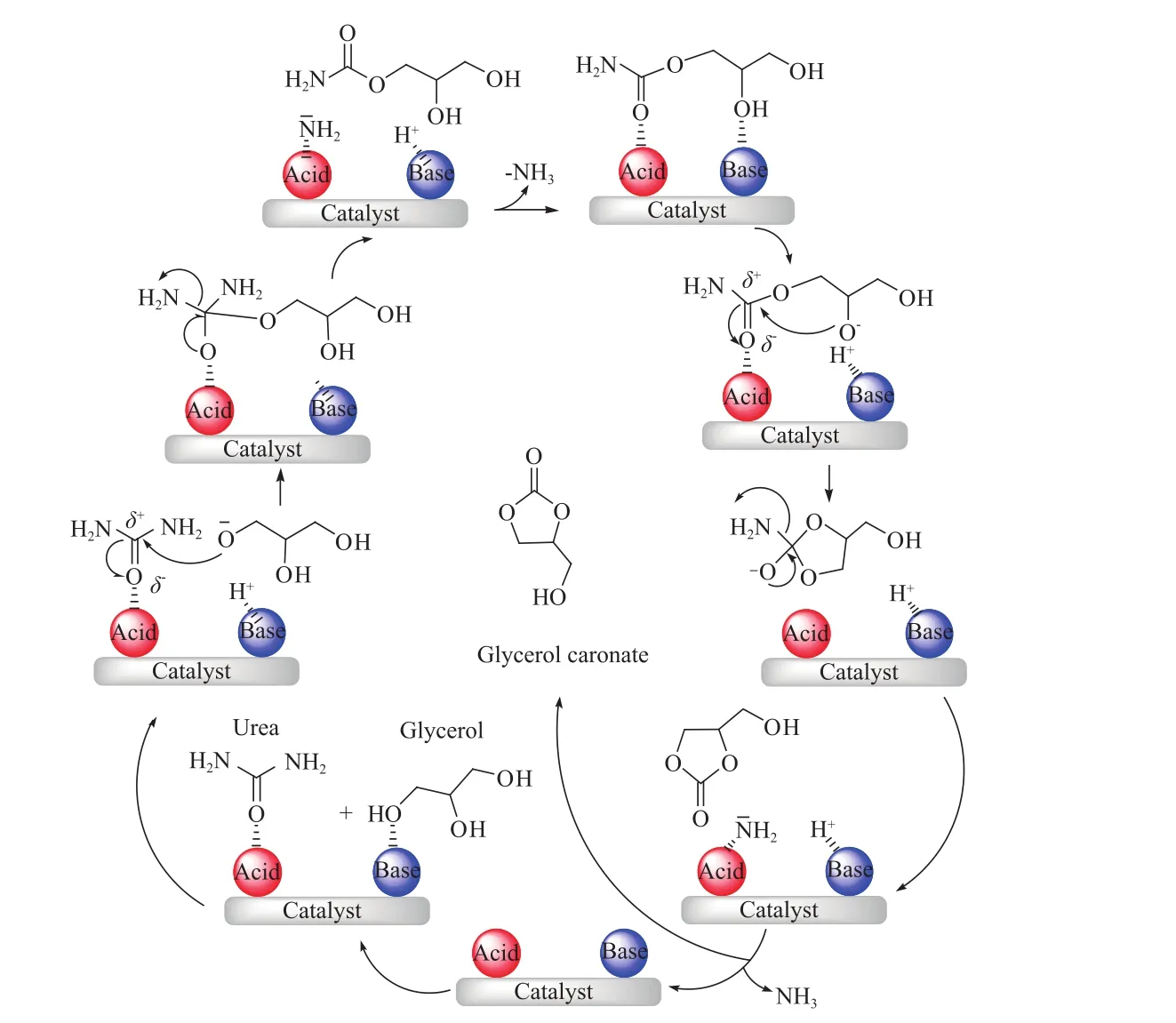
圖1 甘油與尿素的反應機理Fig.1 Proposed mechanism for the reaction of urea and glycerol.
4 結語
隨著GC作為多官能化合物在各個領域需求量的增加,GC的有效制備越來越引起人們的重視。目前GC的幾種主要合成方法均存在缺陷,而尿素與甘油反應合成GC具有反應條件溫和、操作安全及產物產率高等優點。從生態學的角度看,具有環境友好、資源有效利用的甘油與尿素反應路線受到學術界及工業界的極大關注。
在理解文獻及研究的基礎上,從反應機理及實用角度出發,尿素與甘油反應合成GC的高效催化劑研究應該著眼于功能性多相催化劑。該催化劑應具有可以活化甘油羥基的堿性位點及活化尿素羰基的酸性位點,酸性催化活性位點與堿性催化活性位點應盡可能靠近,從而有效活化尿素和甘油,提高反應效率。催化劑以比表面積大的層狀結構、多孔結構為佳,或負載MPR-ILs。只有設計多相催化劑的結構,調節催化劑中的酸堿催化活性中心的催化活性,研究催化劑的構效關系,才可能開發出具有工業應用前景的高效催化劑,并運用到環境友好的GC制備工藝中。
[1] Anitha M,Kamarudin S K,Ko fl i N T. The potential of glycerol as a value-added commodity[J].Chem Eng J,2016,295:119-130.
[2] Wang Jinglun,Yong Tianqiao,Yang Jianwen,et al. Organosilicon functionalized glycerol carbonates as electrolytes for lithium-ion batteries[J].RSC Adv,2015,5 (23):17660-17666.
[3] Joser O G,Olga G J A,Camilo R L,et al. A brief review on industrial alternatives for the manufacturing of glycerol carbonate,a green chemical[J].Org Process Res Dev,2012,16 (3):389-399.
[4] Sonnati M O,Sonla A,Elisabeth P,et al. Glycerol carbonate as a versatile building block for tomorrow:Synthesis,reactivity,properties and applications[J].Green Chem,2013,15 (2):283-306.
[5] 康麗娟,趙新強,安華良,等. CaO-PbO催化酯交換合成甘油碳酸酯[J].石油化工,2011,40 (2):140-145.
[6] Hammond C,Sanchez J A L,Rahim M H A,et al. Synthesis of glycerol carbonate from glycerol and urea with gold-based catalysts[J].Dalton Trans,2011,40 (15):3927-3937.
[7] Shukla K,Srivastava V C. Synthesis of organic carbonates from alcoholysis of urea:A review[J].Catal Rev,2017,59(1):1-43.
[8] Teng W K,Ngon G C,Yusoff R,et al. A review on the performance of glycerol carbonate production via catalytic transesterification:Effects of influencing parameters[J].Energy Convers Manage,2014,88:484-497.
[9] 郭爽,李金麗,王家喜,等. 尿素與甘油反應制甘油碳酸酯的綠色合成工藝[J].化工進展,2012,31 (3):658-661.
[10] 河北工業大學. 一種甘油碳酸酯的制備方法:102285957 A[P].2011-12-21.
[11] Park J H,Choi J S,Woo S K,et al. Isolation and characterization of intermediate catalytic species in the Zn-catalyzed glycerolysis of urea[J].Appl Catal,A,2012,433/434(12):35-40.
[12] Wang Dengfeng,Zhang Xuelan,Liu Chunli,et al. Synthesis of glycerol carbonate from glycerol and urea over lanthanum compounds[J].React Kinet Mech Cat,2015,115 (2):597-609.
[13] Kumar C R,Jagadeeswaraiah K,Prasad P S S,et al. Samarium-exchanged heteropoly tungstate:An efficient solid acid catalyst for synthesis of glycerol carbonate from glycerol and benzylation of anisole[J].ChemCatChem,2012,4 (9):1360-1367.
[14] Jagadeeswaraiah K,Kumar C R,Prasad P S S,et al. Incorporation of Zn2+ions into the secondary structure of heteropoly tungstate:Catalytic efficiency for synthesis of glycerol carbonate from glycerol and urea[J].Catal Sci Technol,2014,4( 9):2969-2977.
[15] Babu M S,Srivani A,Parameswaram G,et al. Understanding the role of tantalum in heteropoly tungstate catalysts for the synthesis of glycerol carbonate from glycerol and urea[J].Catal Lett,2015,145( 9):1784-1791.
[16] Chen Juanjuan,Wang Chang,Dong Bin,et al. Ionic liquids as eco-friendly catalysts for converting glycerol and urea into high value-added glycerol carbonate[J].Chin J Catal,2015,36( 3):336-343.
[17] Turney T W,Patti A,Gates W,et al. Formation of glycerol carbonate from glycerol and urea catalysed by metal monoglycerolates[J].Green Chem,2013,15( 7):1925-1931.
[18] Marcos F R,Casilda V C,Banares M A,et al. Novel hierarchical Co3O4/ZnO mixtures by dry nanodispersion and their catalytic application in the carbonylation of glycerol[J].J Catal,2010,275( 2):288-293.
[19] Casilda V C,Mul G,Fernandez J F,et al. Monitoring the catalytic synthesis of glycerol carbonate by real-time attenuated total reflection FTIR spectroscopy[J].Appl Catal,A,2011,409/410( 23):106-112.
[20] Wang Liguo,Ma Yubo,Wang Ying,et al. efficient synthesis of glycerol carbonate from glycerol and urea with lanthanum oxide as a solid base catalyst[J].Catal Commun,2011,12(15):1458-1462.
[21] Zhang Juan,He Dehua. Lanthanum-based mixed oxides for the synthesis of glycerol carbonate from glycerol and urea[J].React Kinet Mech Cat,2014,113( 2):375-392.
[22] Baek J,Ryu Y B,Kim M H,et al. Synthesis of glycerol carbonate from biomass glycerol:Focus on the calcination temperature[J].Mater Sci Forum,2012,724:374-377.
[23] Ryu Y B,Baek J H,Kim Y D,et al. Synthesis of glycerol carbonate by transesteri fi cation of glycerol with urea over Zn/Al mixed oxide[J].J Nanosci Nanotechno,2015,15( 1):321-325.
[24] Ryu Y B,Kim J S,Kim K H,et al. Synthesis of Zn/Al mixed-oxide catalyst for carbonylation of glycerol with urea[J].Res Chem Intermed,2015,42( 1):83-93.
[25] Phu H N,Park C Y,Shin E W. Activated red mud-supported Zn/Al oxide catalysts for catalytic conversion of glycerol to glycerol carbonate:FTIR analysis[J].Catal Commun,2016,85:52-56.
[26] Jagadeeswaraiah K,Kumar C R,Prasad P S S,et al. Synthesis of glycerol carbonate from glycerol and urea over tintungsten mixed oxide catalysts[J].Appl Catal,A,2014,469( 2):165-172.
[27] Jagadeeswaraiah K,Kumar C R,Rajashekar A,et al. The role of tungsten oxide species supported on titania catalysts for the synthesis of glycerol carbonate from glycerol and urea[J].Catal Lett,2016,146( 3):692-700.
[28] Pandian M,Ravishankar R,Shanbhag G V. Novel bifunctional Zn-Sn composite oxide catalyst for the selective synthesis of glycerol carbonate by carbonylation of glycerol with urea[J].ChemCatChem,2016,8( 3):631-639.
[29] Kim D W,Park M S,Selvaraj M,et al. Catalytic performance of polymer-supported ionic liquids in the synthesis of glycerol carbonate from glycerol and urea[J].Res Chem Intermed,2011,37( 37):1305-1312.
[30] Kim D W,Kim M J,Roshith K,et al. Comparative catalytic activity of supported ZnBr2-containing ionic liquid catalysts for preparation of glycerol carbonate by glycerolysis of urea[J].Korean J Chem Eng,2014,31( 6):972-980.
[31] Kim D W,Park K A,Kim M J,et al. Synthesis of glycerol carbonate from urea and glycerol using polymer-supported metal containing ionic liquid catalysts[J].Appl Catal,A,2014,473( 5):31-40.
[32] Kim D W,Park D W. Organic-inorganic hybrids of imidazole complexes of zinc(Ⅱ) for catalysts in the glycerolysis of urea[J].J Nanosci Nanotechno,2014,14( 6):4632-4638.
[33] Kondawar S E,Potdar A S,Rode C V. Solvent-free carbonylation of glycerol with urea using metal loaded MCM-41 catalysts[J].RSC Adv,2015,5( 21):16452-16460.
[34] Marakatti V S,Halgeri A B. Metal ion-exchanged zeolites as highly active solid acid catalysts for the green synthesis of glycerol carbonate from glycerol[J].RSC Adv,2015,5(19):14286-14293.
[35] Narkhede N,Patel A. Synthesis of glycerol carbonate via glycerolysis of urea catalysed by silicotungstates impregnated to MCM-41[J].RSC Adv,2015,5( 65):52801-52808.
[36] Climent M J,Corma A,Frutos P D,et al. Chemicals from biomass:Synthesis of glycerol carbonate by transesterification and carbonylation with urea with hydrotalcite catalysts:The role of acid-base pairs[J].J Catal,2010,269( 1):140-149.
[37] Sun Yongfa,Tong Xinli,Wu Zhidong,et al. A sustainable preparation of glycerol carbonate from glycerol and urea catalyzed by hydrotalcite-like solid catalysts[J].Energy Technol,2014,2( 3):263-268.
[38] Sandesh S,Shanbhag G V,Halgeri A B,et al. Zinc hydroxystannate:A promising solid acid-base bifunctional catalyst[J].RSC Adv,2013,4( 2):974-977.
[39] Lee S D,Park G A,Kim D W,et al. Catalytic performance of functionalized IRMOF-3 for the synthesis of glycerol carbonate from glycerol and urea[J].J Nanosci Nanotechno,2014,14( 6):4551-4556.
[40] Indran V P,Zuhaimi N A S,Deraman M A,et al. An accelerated route of glycerol carbonate formation from glycerol using waste boiler ash as catalyst[J].RSC Adv,2014,4(48):25257-25267.
[41] Indran V P,Dauda A S H,Maniam G P,et al. Versatile boiler ash containing potassium silicate for the synthesis of organic carbonates[J].RSC Adv,2016,6( 41):34877-34884.
[42] Indran V P,Saud A S H,Maniam G P,et al. Viable glycerol carbonate synthesis through direct crud glycerol utilization from biodiesel industry[J].Waste Biomass Valori,2016,8(4):1049-1059.
[43] Zuhaimi N A S,Indran V P,Deraman M A,et al. Reusable gypsum based catalyst for synthesis of glycerol carbonate from glycerol and urea[J].Appl Catal,A,2015,502:312-319.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
